Fabrication of SERS-fluorescence dual modal nanoprobes and application to multiplex cancer cell imaging†
Sangyeop
Lee
a,
Hyangah
Chon
a,
Soo-Young
Yoon
b,
Eun Kyu
Lee
c,
Soo-Ik
Chang
d,
Dong Woo
Lim
a and
Jaebum
Choo
*a
aDepartment of Bionano Engineering, Hanyang University, Ansan, 426-791, South Korea. E-mail: jbchoo@hanyang.ac.kr
bDepartment of Laboratory Medicine, Korea University College of Medicine, Ansan, 425-707, South Korea
cCollege of Bionano Technology, Kyungwon University, Sungnam, 461-701, South Korea
dDepartment of Biochemistry, Chungbuk National University, Cheongju, 361-763, South Korea
First published on 11th November 2011
Abstract
We report a highly sensitive optical imaging technology using surface-enhanced Raman scattering (SERS)-fluorescence dual modal nanoprobes (DMNPs). Fluorescence microscopy is a well-known imaging technique that shows specific protein distributions within cells. However, most currently available fluorescent organic dyes have relatively weak emission intensities and are rapidly photo-bleached. Thus more sensitive and stable probes are needed. In this work we develop DMNPs, which can be used for both SERS and fluorescence detection. SERS detection is a powerful technique that allows ultrasensitive chemical or biochemical analysis through unlimited multiplexing and single molecule sensitivity. Combining advantages of fluorescence and SERS allows these dual modal nanostructures to be used as powerful probes for novel biomedical imaging. In this work, the fabrication and characterization of the SERS-fluorescence DMNPs and application to biological imaging were investigated using markers CD24 and CD44, which are co-expressed in MDA-MB-231 breast cancer cells, as a model system. SERS imaging with DMNPs was found to be a powerful tool to determine the co-localization of CD24 and CD44 in the cell.
1. Introduction
Highly sensitive optical imaging technology using metal nanoparticles has been extensively applied to cellular imaging and biomedical diagnostics.1–13 At the nanoscale, the surface-to-volume ratios of nanoparticles become large and their electronic energy states become discrete, leading to unique optical, electronic, and mechanical properties of nanoparticles. For example, inorganic metal nanocrystals called quantum dots (QDs) are extensively used as fluorescence labeling agents for biological cells and tissues. QDs offer significant advantages over conventional organic dyes such as brighter fluorescence and higher resistance to photobleaching that make them suitable for ultrasensitive imaging.14–20 Despite their potential success as bioimaging agents, QDs have some intrinsic problems such as cytotoxicity issues and broad emission profiles that lead to band overlapping. These problems have been obstacles to their in vivo and multiplex biomedical applications.6Surface-enhanced Raman scattering (SERS) as the inelastic light scattering of gold or silver nanoparticles is emerging as a powerful alternative for identifying specific biological targets in live cells.21–33 Using individual spectral signatures or their combinations, SERS allows ultra-sensitive biochemical analysis with multiplex targeting.32–35SERS has a detection sensitivity that is 10–14 orders of magnitude higher than conventional Raman spectroscopy, which is comparable with fluorescence detection.23,36–38 Furthermore, with signals that are much narrower than fluorescence emission bands, SERS also allows detection of multiple analytes.15,37 Until now, various types of SERS nanoprobes have been developed for single or multiplex in vitrobiomarker assays using microwell-based gold patterned arrays or lab on a chip-based optofluidic sensors.39–41 However, a few studies have reported on simultaneous detection of multiple biomarkers expressed in live cells using SERS imaging.
Here, we applied the SERS imaging technique to the highly sensitive detection of dual breast cancer markers co-expressed in live cells. For this purpose, silica-encapsulated gold nanoparticles (GNPs) with different Raman reporters were prepared. GNPs are most widely used as optical enhancing agents because of their long-term stability, easily controllable size distribution, and high homogeneity.25,31 Nevertheless, when unprotected GNPs are used in the SERS based cellular imaging, these reporters are easily influenced by their surroundings. When unprotected SERS reporters were used for SERS imaging, not only the SERS signal from reporter molecules, but also the signal from cell components is observed.
Another problem of unprotected GNPs is the lack of reproducibility that results from uncontrolled aggregation. Although the aggregation of nanoparticles substantially enhances SERS signals,42–47 it is very difficult to control in living cells. Therefore, SERS reporters must be prepared with a protective shell that can prevent desorption of Raman reporters as well as adsorption of external species. This is because the reproducibility of nanoparticles is the most important factor for determining accurate protein co-expression and localized spatial distributions of dual biomarkers in living cells. Of various encapsulation materials such as biomolecules, polymers and silica,29,32,48–50silica nanoparticles are superior because of their remarkable stability in salt media and organic solvents and their impermeability to dye molecules. Silica encapsulation greatly enhances the stability of nanoparticles even though their SERS signal intensity is weaker than aggregated nanoparticles. Silica-encapsulated nanoparticles can be used as multiplex bioimaging agents since particles can be labeled with different Raman reporter molecules. Silica-encapsulated nanoprobes have been used for simultaneous imaging of multiple cancer markers expressed in live cells. Recently, Irudayaraj et al.35 reported an efficient detection SERS platform to achieve dual marker detection for three different types of cancer cells. In their work, hot spots were generated by gold nanoparticle networks using DNA hybridization on cell surface marker sites. This network structure is reversible and the signal enhancement during formation could be dynamically monitored in a multiplex format for three different cancer cell types. However, in spite of the excellent multiplex capability of SERS detection, its slow imaging speed is a major hurdle for the fast recognition of specific markers expressed on a cell.
To resolve this problem, fluorescence-SERS dual modal nanoprobes (DMNPs) were fabricated in this study. Silica-encapsulated SERS nanoparticles were labeled with organic dyes for faster fluorescence-based tracking and imaging. Fig. 1 displays the schematic outline for targeting antibody-conjugated DMNPs to specific cancer markers expressed in live cells. The fluorescence signal can be used as a fast detection tool for cancer marker recognition, while the SERS signals can be used as an accurate sensing tool for imaging localized marker distributions.32,51 The thickness of silica was adjusted to 20 nm to prevent fluorescence quenching. An additional encapsulating silica layer was added to protect the fluorescence dyes. For cellular imaging, markers CD24 and CD44, which are co-expressed in MDA-MB-231 breast cancer cells, were the model system. Subpopulations of CD24 and CD44 markers on the cell surface are reported to determine the invasive and metastatic property of tumors. The CD44/CD24 phenotype is associated with enhanced invasive properties and elevated expression of genes involved in invasion. Especially, the strong expression of CD44 plays a critical role in numerous types of human cancers. CD44 is involved in cell differentiation, adhesion, and metastasis of cancer cells.52,53 Fluorescence-SERS DMNPs showed outstanding stability under different pH and salt conditions. This is the first report on the simultaneous fluorescence-SERS imaging technique for duplex co-expressed markers on cancer cells using highly stable silica-encapsulated nanoparticles. This analytical technique is expected to be a powerful tool for understanding localized spatial distributions of cancer markers in living cells, and can be directly applied to early cancer diagnosis.
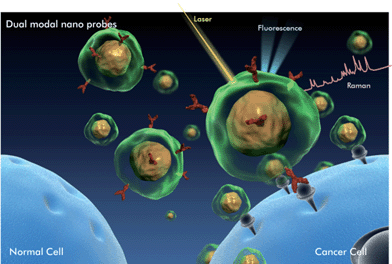 | ||
| Fig. 1 Illustration of cancer marker detection using fluorescence-SERS dual modal nanoprobes (DMNPs). Functional DMNPs are selectively targeted onto cancer markers expressed in live cells. Fluorescence and Raman signals are emitted simultaneously. Fast tracing is possible through fluorescence detection, and Raman detection provides detailed information about molecular interactions. | ||
2. Experimental
2.1. Preparation of SERS active GNPs
GNPs were synthesized using the citrate-reduction method reported by Frens,54 with 0.5 mL of 38.8 mM sodium citrate solution added to 50 mL of 0.294 mM HAuCl4 solution under stirring. After the mixture was boiled for 10 min, heating was stopped and the solution was stirred for 1 h. The resulting solution was cooled to room temperature and stored at 4 °C. UV/Vis spectroscopy and transmission electron microscopy (TEM) were used to identify the average size of gold nanoparticles. Our measurements estimated the average diameter of gold particles to be 40 nm. To prepare SERS active probes as Raman reporters, 3 μL of 10−4 M of malachite green isothiocyanate (MGITC) and 3 μL of 10−4 M of tris(2,2′-bipyridyl)ruthenium(II) chloride hexahydrate (Rubpy) were added to 3 mL of gold nanocolloids. The number of adsorbed reporter molecules per particle was estimated to be approximately 6.8 × 102. After 10 min, mixtures were rinsed with centrifugation at 3000 rpm for 12 min, and precipitates were resuspended in 4 mL of deionized (DI) water.2.2. Silica encapsulation and fluorescence labeling
To prevent aggregation and fluorescence quenching, SERS active gold nanoprobes were encapsulated in a silica spacer shell. Under stirring, 3 mL of SERS colloid probe was diluted with 20 mL of isopropanol, followed by the addition of 200 μL of 28% NH4OH and 5 μL of 10−4 M tetraethyl orthosilicate (TEOS). The reaction was allowed to proceed for 24 h at room temperature. Silica-encapsulated SERS active probes were collected by centrifugation at 3000 rpm for 12 min, and dispersed in 23 mL of isopropanol. To provide SERS-active nanoprobes with fluorescent features, (3-aminopropyl)trimethoxysilane (APTMS) was reacted by mixing 300 μL of 10−4 M with 10 μL of 10−2 M ITC-modified fluorophores (FITC or RuITC) and vortexed for 24 h at room temperature. The isothiocyanate group of FITC and RuITC was covalently bonded to the amine group of APTMS.Finally, SERS-active probes were encapsulated by adding 0.2 μL of the APTMS fluorophore to 1 mL of silica-coated SERS probes. After 30 min, the second silica shell formed around the fluorophore and 0.4 μL of 10−4 M TEOS and 2 μL of 28.8% NH4OH were added. To remove unreacted fluorophores, the mixture was centrifuged at 3000 rpm for 12 min and the precipitate was resuspended in 0.1 mL of DI water.
2.3. Antibody conjugation onto DMNPs
To selectively attach DMNPs to cancer markers on cells, CD24 and CD44 antibodies were conjugated to the two different types of DMNPs. The surface of the silica shell was modified with amine groups by reacting 2 μL of 10−4 M APTMS with 300 μL of nanocolloids for 30 min. This solution was centrifuged at 3000 rpm for 12 min and the supernatant removed. After washing with DI water, the final precipitates were redispersed in DI water and 3 μL of 0.5% glutaraldehyde was added to 1.5 μL of 10 μg mL−1antibody to make amine-functionalized nanoprobes. Finally, the mixture was incubated for 1 h under mild vortexing. In this process, the glutaraldehyde was used as a linker between the antibodies on probe surfaces and the proteins on the cell membranes. After centrifugation, the final solution was suspended in 300 μL of DI water.2.4. Cell preparations
MDA-MB-231 cells were used as the optical imaging target for testing the duplex imaging capability of fluorescence-SERS dual modal probes since the markers CD44 and CD24 are co-expressed on their membranes. MDA-MB-231 cells, seeded on cover glasses, were grown without doxycycline. After 2 days, the cells were fixed with 3.7% paraformaldehyde for 15 min and washed with PBS for 3 times. Then cells were incubated with antibody-conjugated DMNPs for 1.5 h at room temperature.2.5. Fluorescence and Raman measurements
Fluorescence images of DMNP-labeled cells were collected with a Leica TCS SL confocal fluorescence microscope with a 40× (HCX APO U-V-I, NA 1.30) oil immersion objective lens. They were viewed with 488 nm laser excitation, and the emitted fluorescence light was detected between λ = 595 and 635 nm for red probes, and λ = 500 and 540 nm for green probes. Raman measurements were performed with a Renishaw 2000 confocal Raman microscope system. A He–Ne laser operating at λ = 633 nm was used as the excitation source with a laser power of 15 mW. All Raman spectra were obtained with a 10 s exposure time. The Rayleigh line was removed from the collected Raman scattering using a holographic edge filter in the collection path. A charge-coupled device camera was coupled to a spectrograph that, in combination, provided a 2 cm−1 spectral resolution. Raman images were obtained using a Raman point-mapping method, and the exposure time was 1 s.3. Results and discussion
3.1. Fabrication of DMNPs
Fig. 2 shows the sequential process for fabricating SERS-fluorescence DMNPs. First, 40 nm GNPs were synthesized (a) and Raman reporter molecules were adsorbed onto their surface (b). Second, Raman reporter-labeled GNPs were encapsulated with silica to provide a space for further labeling (c). The resulting thin silica spacer prevented the release of Raman reporter molecules and prevented fluorescence quenching from GNPs. The thickness of the silica shell was controlled by changing amounts of TEOS and ammonia to obtain maximum fluorescence intensity. According to our fluorescence measurements, the most intense fluorescence intensity was obtained at a shell thickness of 20 nm. Third, fluorescent dye-coupled APTMS was covalently attached onto their surface (d). Finally, another thin silica layer was encapsulated to minimize nanoparticle aggregations as well as to protect the nanoparticle cores from degradation in subsequent steps (e).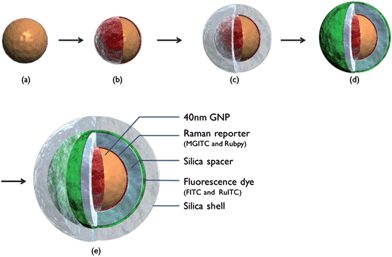 | ||
| Fig. 2 Sequential procedure for fabricating SERS-fluorescence DMNP. (a) 40 nm GNPs; (b) Raman reporter-labeled GNP; (c) inner silica-encapsulated SERS nanoprobe; (d) fluorescent dye-labeled SERS nanoprobe; and (e) outer silica-encapsulated DMNP. | ||
Fig. 3(a) shows TEM images of silica-encapsulated SERS-fluorescence DMNPs. Dynamic light scattering (DLS) measurements were also performed to confirm the size of nanoparticles (Fig. S1†). Here, the core gold particle size was estimated to be 40 ± 3.5 nm. The silica shell thickness could easily be controlled by changing the amount of TEOS. The upper panel in Fig. 3(b) shows 40 nm GNPs (Fig. 2(a)), with silica-encapsulated SERS nanoprobes (Fig. 2(c)), and silica-encapsulated SERS-fluorescence DMNPs (Fig. 2(e)). Only the DMNPs showed bright green fluorescence under UV/Vis irradiation, as shown in the lower panel in Fig. 3(b). Raman reporters are light-emitting, but their fluorescence signal was completely quenched by the gold nanoparticles. However, when the fluorescent dyes were between two silica layers, they displayed a strong fluorescence emission because the first silica layer provided enough distance to prevent fluorescence quenching. The optimal shell thickness for the maximum fluorescence was estimated to be about 20 nm. Fig. S2† shows a variation of the fluorescence intensity along the silica shell thickness.
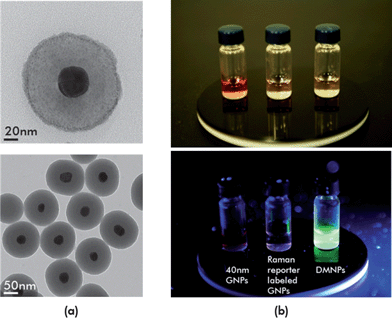 | ||
| Fig. 3 (a) Transmission electron microscopic images of silica-encapsulated SERS-fluorescence DMNPs; (b) fluorescence emission images for GNP colloid, Raman reporter-labeled GNP colloid, and DMNP colloid. UV/Vis irradiation of each sample caused only the DMNP colloid displays to fluoresce because the outer silica shell provided enough distance to prevent fluorescence quenching. | ||
3.2. Chemical stability of DMNPs
An important issue for imaging nanoprobes is stability under different pH and salt conditions. Because the human body is 0.9% saline and pH distributions differ by organ, the stability of nanoprobes in salinity and acidity is crucial for in vivo applications. Fig. S3† shows the stability of SERS-fluorescence DMNPs under different salt and pH conditions. In 0.3% salt solution, unencapsulated GNPs began to aggregate but DMNPs showed no change at higher concentrations of salt, as shown in Fig. S3(a)†. The color of unencapsulated GNPs immediately changed from red to violet, indicating particle aggregation with increased salt concentration. Controlling the degree of aggregation for reproducible imaging analysis is nearly impossible, so aggregation leads to poor homogeneity of SERS signals. At higher concentrations of salt solution (1% and 2%), precipitation of aggregated nanoprobes was observed for unencapsulated GNPs. In contrast, DMNPs showed no critical color change up to 2% salt solution. DMNPs were also very stable to pH changes, as shown in Fig. S3(b)†. However, the color of the unencapsulated GNPs changed at different pH, meaning that aggregation occurred with pH changes.The optical stabilities of DMNPs were tested. A serious problem in biological imaging is photobleaching of fluorophore molecules. Photobleaching occurs when a fluorophore loses the capability to fluoresce because of photon-induced chemical damage and covalent modification, altering the long fluorescence lifetime. The DMNPs resolved the photobleaching problem. To verify photobleaching, human breast cancer cells (MDA-MB-231) were labeled with organic dyes and DMNPs and their fluorescence intensity changes monitored. As shown in Fig. 4(a), FITC-labeled cancer cells lost fluorescence intensity rapidly but the DMNP-labeled cancer cells retained fluorescence intensity for 25 min. This was because the fluorescent molecules spent less time in the excited state, preserving fluorescence intensity without photobleaching. Fig. 4(b) also shows the long-term stability of the DMNP Raman signals. SERS intensity was unchanged for 5 months because the silica shell prevented desorption of Raman reporter molecules as well as adsorption of external species. Another advantage of DMNPs is their signal reproducibility. As shown in Fig. 4(c), the aggregation of unencapsulated GNPs induced SERS signal enhancement in early stages, but this decreased dramatically over time. This indicated that the SERS intensity of unencapsulated GNPs was initially increased by formation of hot spots but decreased rapidly from precipitation by aggregation. The SERS intensity of DMNPs was very consistent with time, however, since the silica encapsulation guaranteed reproducible SERS signals on a particle-to-particle basis. Thus, experimental data on salt concentration, pH, photobleaching, and aggregation showed that the synthesized DMNPs were optically stable under many conditions.
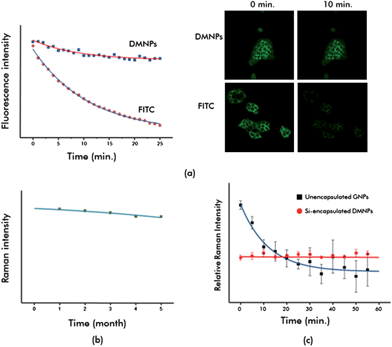 | ||
| Fig. 4 Optical stability test for SERS-fluorescence DMNPs. (a) Photobleaching test for FITC- and DMNP-labeled MDA-MB-231 breast cancer cells; (b) long-term stability test for the DMNP Raman signal; and (c) signal reproducibility test of SERS intensity for unencapsulated GNPs and silica-encapsulated DMNPs over time. | ||
3.3. Evaluation of quantitative spectral multiplexing of DMNPs
An important advantage of SERS nanoprobes is their ability to simultaneously detect multiple targets. In our assays, two different Raman reporter-labeled DMNPs (MGITC and Rubpy), combined with two different fluorescence dyes (FITC, green; RuITC, red) were prepared to evaluate multiplexing capability. Fig. S4† shows duplex fluorescence images (a) and SERS spectra for dual modal nanoprobes (b), with (a,b) and (c,d) sets having the same fluorescence emission colors (green and red) but showing different Raman spectra. The (a,c) and (b,d) sets show the same Raman spectra but different fluorescence emission colors. This indicated that the fluorescence features could be controlled by the dyes in the second silica layer regardless of the Raman reporters adsorbed onto the first layer. This was also valid for the Raman features.Quantitative multiplexing capability of DMNPs was evaluated using a mixture of two probes at different ratios. Fig. 5(a) displays the variation of Raman spectra for different volume ratios of two Raman reporter-labeled GNPs (MGITC and Rubpy). Intensity variations of the strongest Raman peak (1615 cm−1 for MGITC and 1485 cm−1 for Rubpy) were used for quantitative analysis. As shown in Fig. 5(b), the variation in peak intensity as a function of molar ratio between MGITC and Rubpy-labeled GNPs was excellent at five different ratios.
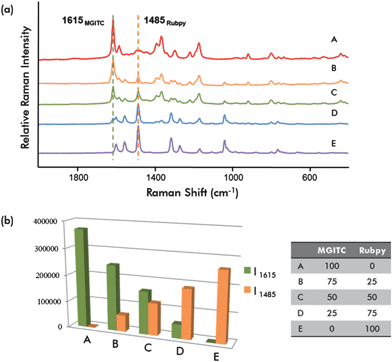 | ||
| Fig. 5 (a) SERS spectra for different molar ratios of DMNP mixtures. The strongest Raman peaks at 1615 cm−1 (MGITC) and at 1485 cm−1 (Rubpy) were used for quantitative analysis. (b) Peak intensity variation as a function of the molar ratio of MGITC and Rubpy-labeled GNPs. | ||
3.4. Duplex imaging of dual markers expressed in cancer cells using DMNPs
The fluorescence signal can be used as an immediate indicator of molecular recognition, while the SERS signal can be used as a signature of specific molecular interactions. To validate the multiplex detection capability of DMNPs, fluorescence and SERS images for co-expressed cancer markers were measured using a confocal laser-induced fluorescence scanning microscope and a confocal Raman microscope. First, two different Raman/fluorescence combination DMNPs (Rubpy/RuITC and MGITC/FITC) were prepared for SERS-fluorescence dual imaging. RuITC and FITC have fluorescence emissions at 615 nm and 518 nm, respectively. Rubpy and MGITC have the strongest Raman peaks at 1485 cm−1 and 1618 cm−1. For more specific targeting, anti-CD24 and anti-CD44 were covalently bound to the surface of RuITC/Rubpy and FITC/MGITC DMNPs by typical immunohistochemistry protocols. Fig. S5† shows the sequential procedure for antibody conjugation onto the DMNP surface. Glutaraldehyde was used to link antibodies to the probe surface.For fluorescence-SERS imaging of dual cancer markers in a single cell, CD24- and CD44-expressing MDA-MB-231 breast cancer cells were prepared and imaged after 1.5 h of incubation with two different types of DMNPs: anti-CD24 conjugated Rubpy/RuITC DMNPs and anti-CD44 conjugated MGITC/FITC DMNPs. Rubpy/RuITC and MGITC/FITC DMNPs successfully bound to CD24 and CD44 and showed their locations on the cell.
Fig. 6 shows confocal fluorescence scanning images and Raman mapping images for dual marker-expressing MDA-MB-231 cells after incubation with DMNPs. Fig. 6(a) and (b) show fluorescence images of RuITC and SERS from Rubpy reporters for CD24 marker distribution. Fig. 6(c) and (d) show fluorescence images from FITC and the SERS signals from MGITC for CD44 distribution.
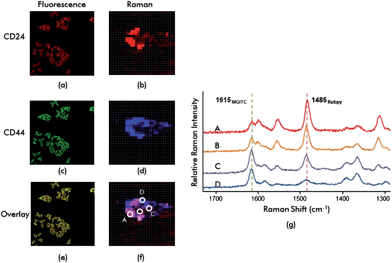 | ||
| Fig. 6 Fluorescence-SERS duplex imaging of CD24 and CD44 markers expressed in MDA-MB-231 breast cancer cells. (a) Fluorescence and (b) SERS images for CD24 markers; (c) fluorescence and (d) SERS images for CD44 markers; (e) fluorescence and (f) SERS overlay images for CD24 and CD44 markers; (g) Raman spectra for four different spots (A, B, C and D) for the cell in (f). The 1485 cm−1 peak of Rubpy and 1618 cm−1 peak of MGITC were used to identify localized distribution of CD24 and CD44 markers in the cell. | ||
Fig. 6(e) and (f) show overlays of fluorescence and SERS for distributions of CD24 and CD44. Non-overlapping areas in Fig. 6(e) indicate that both markers are uniquely distributed. Fig. 6(f) also shows non-overlap between red and blue for Raman mapping images. This indicated that the distributions of the two cancer markers CD24 and CD44 were different. The distributions of CD24 and CD44 were clearly identified from Raman spectra. Fig. 6(g) shows Raman spectra for four different points (A, B, C and D) for the cell shown in Fig. 6(f). The 1485 cm−1 peak of Rubpy and 1618 cm−1 peak of MGITC are unique and non-overlapping candidates for identifying specific reporter molecules. From the spectral analysis of all spots, we found that both the CD24 and CD44 markers were equally distributed in the B and C spots because the intensities at 1485 cm−1 and 1618 cm−1 peak were equally strong. We presume that the localized distribution of CD24 was predominant over CD44 at the A spot since the peak intensity at 1485 cm−1 was stronger than the intensity at 1618 cm−1, and vice versa at the D spot. This suggested that the functionalized DMNPs could be used as probes for multiplex SERS imaging. Our data showed that the spatial distribution of specific biomarkers in a cancer cell could be determined by mapping the localization of markers.
4. Conclusions
We have demonstrated the potential applicability of DMNPs as fluorescence and SERS sensing probes for targeting and imaging specific cancer markers in living cells. Silica-encapsulated DMNPs showed superior stability over unencapsulated GNPs since protective silica shells prevented desorption of Raman reporters and adsorption of external species. In addition, DMNPs could be used as multimodal sensing agents for both SERS and fluorescence detection. In this study, anti-CD24-conjugated Rubpy/RuITC DMNPs and anti-CD44-conjugated MGITC/FITC DMNPs were made for imaging the local distribution of CD24 and CD44 markers co-expressed on MDA-MB-231 breast cancer cells. Fluorescence imaging was used as a fast indicator of molecular recognition, while SERS imaging was subsequently used to determine the signature of specific molecular interaction. In particular, the SERS imaging technique with functionalized DMNPs made detailed local distribution of CD24 and CD44 possible.Acknowledgements
This work was supported by the National Research Foundation of Korea (grant numbers R11-2009-044-1002 and K20904000004-09A0500-00410).Notes and references
- N. L. Rosi and C. A. Mirkin, Chem. Rev., 2005, 105, 1547–1562 CrossRef CAS.
- X. Michalet, F. F. Pinaud, L. A. Bentolila, J. M. Tsay, S. Doose, J. J. Li, G. Sundaresan, A. M. Wu, S. S. Gambhir and S. Weiss, Science, 2005, 307, 538–544 CrossRef CAS.
- A. M. Smith, H. W. Duan, A. M. Mohs and S. M. Nie, Adv. Drug Delivery Rev., 2008, 60, 1226–1240 CrossRef CAS.
- S. Courty, C. Luccardini, Y. Bellaiche, G. Cappello and M. Dahan, Nano Lett., 2006, 6, 1491–1495 CrossRef CAS.
- G. Ruan, A. Agrawal, A. I. Marcus and S. Nie, J. Am. Chem. Soc., 2007, 129, 14759–14766 CrossRef CAS.
- A. M. Derfus, W. C. W. Chan and S. N. Bhatia, Nano Lett., 2004, 4, 11–18 CrossRef CAS.
- L. Wang, K. M. Wang, S. Santra, X. J. Zhao, L. R. Hilliard, J. E. Smith, J. R. Wu and W. H. Tan, Anal. Chem., 2006, 78, 646–654 CrossRef.
- M. Nakamura, M. Shono and K. Ishimura, Anal. Chem., 2007, 79, 6507–6514 CrossRef CAS.
- K. Aslan, M. Wu, J. R. Lakowicz and C. D. Geddes, J. Am. Chem. Soc., 2007, 129, 1524–1525 CrossRef CAS.
- E. B. Voura, J. K. Jaiswal, H. Mattoussi and S. M. Simon, Nat. Med., 2004, 10, 993–998 CrossRef CAS.
- X. Gao, Y. Cui, R. M. Levenson, L. W. Chung and S. Nie, Nat. Biotechnol., 2004, 22, 969–976 CrossRef CAS.
- L. R. Hirsch, R. J. Stafford, J. A. Bankson, S. R. Sershen, B. Rivera, R. E. Price, J. D. Hazle, N. J. Halas and J. L. West, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 13549–13554 CrossRef CAS.
- I. H. El-Sayed, X. H. Huang and M. A. El-Sayed, Nano Lett., 2005, 5, 829–834 CrossRef CAS.
- J. K. Jaiswal, H. Mattoussi, J. M. Mauro and S. M. Simon, Nat. Biotechnol., 2002, 21, 47–51.
- M. Bruchez, M. Moronne, P. Gin, S. Weiss and A. P. Alivisatos, Science, 1998, 281, 2013–2016 CrossRef CAS.
- W. C. W. Chan and S. M. Nie, Science, 1998, 281, 2016–2018 CrossRef CAS.
- W. C. W. Chan, D. J. Maxwell, X. H. Gao, R. E. Bailey, M. Y. Han and S. M. Nie, Curr. Opin. Biotechnol., 2002, 13, 40–46 CrossRef CAS.
- C.-y. Zhang and L. W. Johnson, J. Am. Chem. Soc., 2006, 128, 5324–5325 CrossRef CAS.
- S. P. Mulvaney, M. D. Musick, C. D. Keating and M. J. Natan, Langmuir, 2003, 19, 4784–4790 CrossRef.
- M. V. Yezhelyev, A. Al-Hajj, C. Morris, A. I. Marcus, T. Liu, M. Lewis, C. Cohen, P. Zrazhevskiy, J. W. Simons, A. Rogatko, S. Nie, X. Gao and R. M. O'Regan, Adv. Mater., 2007, 19, 3146 CrossRef CAS.
- N. Uzunbajakava, A. Lenferink, Y. Kraan, B. Willekens, G. Vrensen, J. Greve and C. Otto, Biopolymers, 2003, 72, 1–9 CrossRef CAS.
- A. V. Feofanov, A. I. Grichine, L. A. Shitova, T. A. Karmakova, R. I. Yakubovskaya, M. Egret-Charlier and P. Vigny, Biophys. J., 2000, 78, 499–512 Search PubMed.
- J. Kneipp, H. Kneipp, W. L. Rice and K. Kneipp, Anal. Chem., 2005, 77, 2381–2385 CrossRef CAS.
- K. Kneipp, A. S. Haka, H. Kneipp, K. Badizadegan, N. Yoshizawa, C. Boone, K. E. Shafer-Peltier, J. T. Motz, R. R. Dasari and M. S. Feld, Appl. Spectrosc., 2002, 56, 150–154 CrossRef CAS.
- K. Kneipp, H. Kneipp and J. Kneipp, Acc. Chem. Res., 2006, 39, 443–450 CrossRef CAS.
- S. Lee, S. Kim, J. Choo, S. Y. Shin, Y. H. Lee, H. Y. Choi, S. Ha, K. Kang and C. H. Oh, Anal. Chem., 2007, 79, 916–922 CrossRef CAS.
- X.-M. Qian and S. M. Nie, Chem. Soc. Rev., 2008, 37, 912–920 RSC.
- J.-H. Kim, J.-S. Kim, H. Choi, S.-M. Lee, B.-H. Jun, K.-N. Yu, E. Kuk, Y.-K. Kim, D. H. Jeong, M.-H. Cho and Y.-S. Lee, Anal. Chem., 2006, 78, 6967–6973 CrossRef CAS.
- S. Keren, C. Zavaleta, Z. Cheng, A. de la Zerda, O. Gheysens and S. S. Gambhir, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 5844–5849 CrossRef CAS.
- C. L. Zavaleta, B. R. Smith, I. Walton, W. Doering, G. Davis, B. Shojaei, M. J. Natan and S. S. Gambhir, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 13511–13516 CrossRef CAS.
- S. Lee, H. Chon, M. Lee, J. Choo, S. Y. Shin, Y. H. Lee, I. J. Rhyu, S. W. Son and C. H. Oh, Biosens. Bioelectron., 2009, 24, 2260–2263 CrossRef CAS.
- A. Pallaoro, G. B. Braun, N. O. Reich and M. Moskovits, Small, 2010, 6, 618–622 CrossRef CAS.
- M. Xiao, J. Nyagilo, V. Arora, P. Kulkarni, D. S. Xu, X. K. Sun and D. P. Dave, Nanotechnology, 2010, 21, 035101 Search PubMed.
- A. Matschulat, D. Drescher and J. Kneipp, ACS Nano, 2010, 4, 3259–3269 CrossRef CAS.
- K. Lee, V. P. Drachev and J. Irudayaraj, ACS Nano, 2011, 5, 2109–2117 CrossRef CAS.
- S. M. Nie and S. R. Emery, Science, 1997, 275, 1102–1106 CrossRef CAS.
- S. Chan, S. Kwon, T. W. Koo, L. P. Lee and A. A. Berlin, Adv. Mater., 2003, 15, 1595–1598 CrossRef CAS.
- W. E. Doering and S. M. Nie, Anal. Chem., 2003, 75, 6171–6176 CrossRef CAS.
- H. Chon, C. Lim, S. M. Ha, Y. Ahn, E. K. Lee, S. I. Chang, G. H. Seong and J. Choo, Anal. Chem., 2010, 82, 5290–5295 CrossRef CAS.
- J. Choo, H. Hwang, H. Chon and J. K. Park, Anal. Chem., 2010, 82, 7603–7610 CrossRef CAS.
- J. Choo, M. Lee, S. Lee, J. H. Lee, H. W. Lim, G. H. Seong, E. K. Lee, S. I. Chang and C. H. Oh, Biosens. Bioelectron., 2011, 26, 2135–2141 CrossRef.
- C. E. Talley, J. B. Jackson, C. Oubre, N. K. Grady, C. W. Hollars, S. M. Lane, T. R. Huser, P. Nordlander and N. J. Halas, Nano Lett., 2005, 5, 1569–1574 CrossRef CAS.
- F. Svedberg, Z. Li, H. Xu and M. Käll, Nano Lett., 2006, 6, 2639–2641 CrossRef CAS.
- G. H. Gu and J. S. Suh, Langmuir, 2008, 24, 8934–8938 CrossRef CAS.
- Q. Zhou, G. Zhao, Y. Chao, Y. Li, Y. Wu and J. Zheng, J. Phys. Chem. C, 2007, 111, 1951–1954 CrossRef CAS.
- S. Basu, S. K. Ghosh, S. Kundu, S. Panigrahi, S. Praharaj, S. Pande, S. Jana and T. Pal, J. Colloid Interface Sci., 2007, 313, 724–734 CrossRef CAS.
- T. Kim, C. H. Lee, S. W. Joo and K. Lee, J. Colloid Interface Sci., 2008, 318, 238–243 CrossRef CAS.
- D. A. Rey, A. D. Strickland, D. Kirui, N. Niamsiri and C. A. Batt, ACS Appl. Mater. Interfaces, 2010, 2, 1804–1810 Search PubMed.
- M.-A. Woo, S.-M. Lee, G. Kim, J. Baek, M. S. Noh, J. E. Kim, S. J. Park, A. Minai-Tehrani, S.-C. Park, Y. T. Seo, Y.-K. Kim, Y.-S. Lee, D. H. Jeong and M.-H. Cho, Anal. Chem., 2009, 81, 1008–1015 CrossRef CAS.
- K. Kim, Y. M. Lee, H. B. Lee and K. S. Shin, ACS Appl. Mater. Interfaces, 2009, 1, 2174–2180 CrossRef CAS.
- D. C. Kennedy, K. A. Hoop, L. L. Tay and J. P. Pezacki, Nanoscale, 2010, 2, 1413–1416 RSC.
- M. Al-Hajj, M. S. Wicha, A. Benito-Hernandez, S. J. Morrison and M. F. Clarke, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 3983–3988 CrossRef CAS.
- C. Sheridan, H. Kishimoto, R. K. Fuchs, S. Mehrotra, P. Bhat-Nakshatri, C. H. Turner, R. Goulet, S. Badve and H. Nakshatri, Breast Cancer Res., 2006, 8, R59 Search PubMed.
- G. Frens, Nature, Phys. Sci., 1973, 241, 20–22.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1nr11243k |
| This journal is © The Royal Society of Chemistry 2012 |