Nanocrystalline carbonate-apatites: role of Ca/P ratio on the upload and release of anticancer platinum bisphosphonates†
Michele
Iafisco
*ab,
Barbara
Palazzo
a,
Gianmario
Martra
c,
Nicola
Margiotta
d,
Sara
Piccinonna
d,
Giovanni
Natile
d,
Valentina
Gandin
e,
Cristina
Marzano
e and
Norberto
Roveri
a
aDipartimento di Chimica “G. Ciamician”, Alma Mater Studiorum, Università di Bologna,
Via Selmi 2, 40126, Bologna, Italy. E-mail: michele.iafisco@unibo.it; Fax: +390512099593; Tel: +390512099489
bDipartimento di Scienze Mediche, Università del Piemonte Orientale,
Via Solaroli 17, 28100, Novara, Italy
cDipartimento di Chimica IFM & Centro Interdipartimentale di Eccellenza “Nanostructured Interfaces and Surfaces”—NIS Università degli Studi di Torino,
Via P. Giuria 7, 10125, Torino, Italy
dDipartimento Farmaco-Chimico, Università degli Studi di Bari “A. Moro”,
Via Orabona 4, 70125, Bari, Italy
eDipartimento di Scienze Farmaceutiche, Università di Padova,
Via Marzolo 5, 35131, Padova, Italy
First published on 10th November 2011
Abstract
In the present study two nanocrystalline apatites have been investigated as bone-specific drug delivery devices to be used for treatment of bone tumors either by local implantation or by injection. In order to assess how the Ca/P ratio can influence the adsorption and release of anticancer platinum–bisphosphonate complexes, two kinds of apatite nanocrystals having different Ca/P ratios but similar morphologies, degree of crystallinity, and surface areas have been synthesized and characterized. The two platinum–bisphosphonate complexes considered were the bis-{ethylenediamineplatinum(II)}-2-amino-1-hydroxyethane-1,1-diyl-bisphosphonate and the bis-{ethylenediamineplatinum(II)}medronate. The Ca/P ratio plays an important role in the adsorption as well as in the release of the two drugs. In fact, the apatite with a higher Ca/P ratio showed greater affinity for both platinum complexes. Also the chemical structure of the two Pt complexes appreciably affects their affinity towards as well as their release from the two kinds of apatites. In particular, the platinum complex whose bisphosphonate contains a free aminic group showed greater upload and smaller release. The cytotoxicity of the Pt complexes released from the apatite was tested against human cervical, colon, and lung cancer cells as well as against osteosarcoma cells. In agreement with previous work, the Pt complexes released were found to be more cytotoxic than the unmodified complexes.
Introduction
Nanomedicine is an emerging field of biomedical research and it is defined as the application of nanotechnologies for improving healthcare. It takes advantage of the improved and novel physical, chemical, and biological properties of materials at nanoscale dimensions. One of the most promising and studied field in nanomedicine is the development of nanosized drug carriers to deliver anticancer agents to cancerous tissues.1–3 In fact, the functionalities of nanoparticulate delivery systems provide the opportunity: (1) to prolong the circulation in the bloodstream and hence to increase the likelihood of accumulation at tumor sites via the enhanced permeation and retention (EPR) effect, (2) to specifically target to cells or tissues of interest via functionalization with ligands specific for cell surface receptors, (3) to respond to local stimuli in vivo (such as responses to changes in temperature and pH), and (4) to overcome the cell membrane barrier and avoid the enzyme filled lysosomes where degradation occurs.4 Consequently, desired features of the ideal delivery system include: (1) non-toxic starting materials and degradation products, (2) small size (in the range of 10–100 nm) and large surface area for improved uptake, (3) encapsulation of the active agent within the delivery system (as opposed to surface decoration or adsorption where the active agent is not protected from the environment), (4) colloidal stability of the delivery system (to prevent agglomeration in vivo during transport), (5) suitable clearance mechanism (to avoid side effects due to drug loaded particles), (6) long clearance times (to allow adequate time for the delivery system to reach target cells and undergo endocytosis), (7) controlled release of an active agent (such as a pH trigger), and (8) targetability of the delivery system (to enable delivery of particles to cells of choice).5 Moreover, active targeting moieties can also be incorporated into the nano-devices to specifically enhance their internalization by the target cells.4 Most of the ligands used to achieve this aim do so by binding onto specific structures overexpressed by neoplastic cells.6 Subsequent receptor-mediated endocytosis then leads to the uptake of the targeting moiety, along with the nanoparticle and its attached drug payload. Various types of targeting moieties have been used to promote the receptor mediated endocytosis of drug loaded nanoparticles: (1) folic acid, a small molecular weight compound that binds to the membrane receptor overexpressed in a variety of tumors;7 (2) peptides ligands, due to the fact that the receptor of numerous hypothalamic peptides, such as somatostatin, result overexpressed in a variety of neoplastic tissues;8 (3) antibodies, after binding to their cancer-cell-specific membrane antigen most antibodies are internalized by endocytosis.8Many different systems have been investigated as drug delivery carriers, including micelles, vesicles, polymers, microspheres, hydrogels, and solid implantable devices.9Inorganic nanoparticles and, among them, those based on calcium phosphates such as hydroxyapatites, which are the major inorganic components present in tissues of vertebrates,10 have gained particular consideration as materials for nanocarriers.11 Indeed, when compared to other inorganic carriers for drug delivery (silica, quantum dots, carbon nanotubes, and magnetic particles), hydroxyapatites possess the following advantages: favourable biodegradability and biocompatibility, greater solubility and smaller toxicity, low production costs and excellent storage properties (not easily subjected to microbial degradation),12 and pH-dependent dissolution (in particular they dissolve at low pH, e.g. in lysosomes, after cellular uptake or in the acidic environment of several solid tumours, thereby releasing the incorporated drugs or any other biologically active molecule).11,13 Moreover, hydroxyapatite is well known for its capability to bind a wide variety of molecules and therapeutic agents.14,15 Its functionalization with anticancer molecules can render it an innovative biomaterial, able to transfer information and act specifically into a biological environment. In other words, the implanted biomaterial can act not only as a local scaffold for cell invasion and formation of new tissue, but also as a means to deliver previously loaded biomolecules.16,17 In this way apatite nanocrystals can enhance their osteointegration or osteoinduction properties and also stimulate specific cellular responses at a molecular level.
Nowadays cisplatin is one of the five most used drugs in the treatment of solid tumors such as testicular, ovarian, and bladder carcinomas.18 However, the use of cisplatin is limited by some serious drawbacks such as nausea, vomiting, ototoxicity, myelotoxicity and concentration dependent nephrotoxicity.19 The systemic side effects of cisplatin and related Pt drugs can be lowered by several methods. A promising strategy makes use of carrier ligands able to promote a specific accumulation of the drug in the target tissue.20 The local treatment of the tumor by the implantation of a matrix in which the drug has been embedded could represent another appealing possibility. Bisphosphonates (BPs) appear to be promising carrier ligands for targeting a drug to calcium-containing tissues, such as bone.21 They are characterized by two phosphonate groups attached to a single carbon atom and have strong affinity for the Ca2+ ions of bone apatite.22,23 BPs are already clinically used to inhibit demineralization by osteoclasts and to shift the balance between mineralization and demineralization towards bone formation. They not only bind to hydroxyapatite, which is deposited by osteoblasts during bone formation, but are also able to inhibit some cellular mechanisms responsible for bone resorption, such as the inhibition of the enzyme farnesyl diphosphate synthase, which regulates osteoclastic activity.24 On the basis of the previous consideration, we came to the conclusion that platinum–bisphosphonate complexes could be loaded onto calcium-containing matrices to be used as bone fillers. Such composites could be administered locally at the site of an osteosarcoma. In fact, after surgical removal of a bone tumor, a substitute of the lost bone tissue is necessary and materials for bone grafting and replacement have been designed. Moreover, a subsequent antitumor treatment needs to be carried out to avoid recurrence. Therefore, a bioactive and drug releasing material could perform both these features.
In this context we have investigated hydroxyapatite nanocrystals loaded with platinum drugs to be used as bone substitutes able to release an antitumor agent. The final goal is to locally inhibit the tumor re-growth and reduce the systemic toxicity.
In previous works different kinds of biomimetic apatite nanocrystals, having different crystal shapes and chemico-physical properties, were investigated for their ability to adsorb and release cisplatin, alendronate, and two platinum complexes (bis-{ethylenediamineplatinum(II)}-2-amino-1-hydroxyethane-1,1-diyl-bisphosphonate and bis-{ethylenediamineplatinum(II)}medronate).25,26 The results revealed a high loading capacity of the hydroxyapatites for the platinum–bisphosphonate complexes and the ability of the composite material to release the Pt compound to the surrounding medium in a slow kinetic. However, the uptake of platinum complexes was quite similar for the different types of hydroxyapatites and the slightly greater uptake observed for one type of hydroxyapatite was probably due to its larger surface area and disorder, directly related to its lower degree of crystallinity. In addition, the cytotoxicity of Pt–bisphosphonate complexes released from the hydroxyapatite were tested against human cervix carcinoma cells and, interestingly, were found to be more cytotoxic than the unmodified complexes. The released Pt species appears to be the active dichloridodia(m)mineplatinum(II), or related solvato species, formed by dissociation of the bisphosphonate.26
Inspired by the previous works, in the present study we have investigated the adsorption and release of two platinum complexes containing geminal bisphosphonates, differing in their charges and physico-chemical properties, towards two kinds of hydroxyapatite nanocrystals similar in morphology, dimensions, and surface area but differing in the Ca/P ratio. The aim was to unravel how the Ca/P ratio of apatite nanocrystals influences the uptake and the release of platinum–bisphosphonate complexes and how the apatite nanocrystals can perform as bone-specific drug delivery devices to be used for treatment of bone tumours not only upon local implantation but also as injectable materials due to their nano-dimensions.
Experimental
Materials
Common high-purity chemical reagents, MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) and cisplatin were supplied by Sigma Chemical Co. Ultrapure water (0.22 mS, 25 °C) was used in all experiments.Synthesis of platinum–bisphosphonate complexes
Bis-{ethylenediamineplatinum(II)}-2-amino-1-hydroxyethane-1,1-diyl-bisphosphonate (A), bis-{ethylenediamineplatinum(II)}medronate (B), and dichloridoethylenediamineplatinum(II) ([PtCl2(en)], en = ethylenediamine) were prepared as already reported in the literature.25,27 The purity of the Pt complexes was higher than 95% as established by combustion analysis carried out with a Hewlett Packard 185 C, H, and N analyzer. The spectroscopic characterization of the platinum complexes was consistent with literature data. The two dinuclear Pt–bisphosphonate complexes A and B were found to be very stable in aqueous solution (in the conditions used to carry on the present work) even after standing for several months at room temperature, as evidenced by NMR spectroscopy (data not shown).Synthesis of apatite nanocrystals
Apatite nanocrystals were synthesized according to the method reported by Liou et al.28Apatite nanocrystals were precipitated from a suspension of Ca(CH3COO)2 by the slow addition (1 drop s−1) of an aqueous solution of H3PO4, keeping the pH at a constant value of 10 by the addition of a (NH4)OH solution. The reaction mixture was kept under stirring at room temperature for 24 hours, then stirring was suspended and the mixture was left standing for 2 hours to allow deposition of the inorganic phase. This latter was isolated by centrifugation of the reaction mixture, repeatedly washed with water, and freeze-dried at −60 °C under vacuum (3 mbar) overnight.To produce apatites with stoichiometric Ca/P ratio (1.67), the Ca(CH3COO)2 suspension and the H3PO4 solution used were 83 and 50 mM, respectively (the resulting material will be referred to as HA); whereas, to precipitate calcium deficient HA, the Ca(CH3COO)2 suspension and the H3PO4 solution were 75 and 50 mM, respectively (the resulting material will be referred to as CDHA). The HA and CDHA granular fractions having dimensions ranging from 100 to 150 μm were selected for the study.
Characterization of apatite nanocrystals
The specific surface area was determined with a Micromeritics ASAP 2010, by measuring the nitrogen adsorption at 77 K following the BET model (hereafter, SSABET).The Ca/P ratio was determined by inductively coupled plasma-optical emission spectrometry (ICP-OES, Liberty 200, Varian, Clayton South, Australia). Samples were dissolved in 1% wt ultrapure nitric acid. The following analytical wavelengths were chosen: Ca 422 nm, P 213 nm.
The carbonate content was evaluated on dried samples by thermogravimetric analysis (TGA) using a Thermal Analysis SDT Q 600 (TA Instruments, New Castle, DE, USA). Heating was performed in a nitrogen flow (100 mL min−1) using an alumina sample holder at a rate of 10 °C min−1 up to 1200 °C. The weight of the samples was approximately 10 mg.
X-Ray photoemission spectroscopy (XPS) analyses were performed using an M-Probe Instrument (SSI) equipped with a monochromatic Al Kα source (1486.6 eV), having a spot size of 200 × 750 μm and a pass energy of 25 eV, and providing a resolution of 0.74 eV. Using a monochromatic source, an electron flood gun was used to compensate the build up of positive charge on the insulator during the analyses. 10 eV electrons were selected to perform the measurements on these samples. The accuracy of the reported binding energies (BEs) was estimated to be ±0.2 eV. The quantitative data were also accurately checked and reproduced several times (ten times for each sample) and the percentage error was estimated to be ±1%.
The infrared spectra were recorded in the wavelength range from 4000 to 400 cm−1 with 2 cm−1 resolution using a Thermo Nicolet 380 FT-IR spectrometer. A powdered sample (approximately 1 mg) was mixed with about 100 mg of anhydrous KBr. The mixture was pressed at 10 t pressure into 7 mm diameter discs. A pure KBr disk was used as blank. Deconvolution of ν3CO3 and ν4PO4 was performed from FTIR spectra using GRAMS curve-fitting software.
The X-ray diffraction (XRD) patterns of the powders were recorded with an PANalytical X'Pert Pro equipped with an X'Celerator detector powder diffractometer using Cu Kα radiation generated at 40 kV and 40 mA. The instrument was configured with 1/2° divergence and receiving slits. A quartz sample holder was used. The 2θ range was from 5° to 60° with a step size (2θ) of 0.05° and a counting time of 3 s. The degree of HA crystallinity was calculated according to eqn (1):
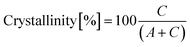 | (1) |
The crystal domain sizes, along the HA axis directions, were calculated applying the Scherrer equation (eqn (2)):
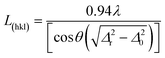 | (2) |
Electrophoretic determinations were performed by a Coulter DELSA apparatus. A Coulter DELSA 440 instrument measured the electrophoretic velocities of suspended particles by measuring the Doppler shift of scattered laser light simultaneously at four different scattering angles: 7.5, 15.0, 22.5 and 30.0°. The suspensions of HA and CDHA were prepared as follows: 0.05 g L−1 of apatite in 10−2 M KNO3 (constant ionic strength), at spontaneously constant pH (6.5).
High resolution transmission electron microscopy (HR-TEM) images of the materials (powder grains dispersed on lacey carbon Cu grids) were obtained with a JEOL 3010-UHR at acceleration potential of 300 kV. As apatite samples might evolve under the electron beam, potentially leading to further crystallisation and/or to a loss of bulk water, observations were carried out under feeble illumination conditions to avoid any modification of the materials during the analysis.29 Fourier transform of direct images was performed by using the DigitalMicrograph program by Gatan.
Determination of equilibrium adsorption isotherms and adsorption kinetics
An aliquot (1.5 mL) of platinum complex solution (ranging from 0.1 to 1 mg mL−1) was added to 5 mg of apatite, to calculate the adsorption isotherm, or to 10 mg of apatite, to calculate the adsorption kinetics, in a 2 mL conical polyethylene Eppendorf tube. After 15 s of treatment in a vortex apparatus, the HA suspension was maintained in a bascule bath at 37 °C. The adsorption profile for the platinum complex was determined by measuring the concentration of Pt complex remaining in the supernatant solution as a function of time. At scheduled times (24 hours for the adsorption isotherm), aliquots (100 μL) of the supernatant (that was well separated from the solid phase by 3 min of centrifugation at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm, 13000g, on a micro Centrifugette 4214) were removed for Pt complex quantification and replaced with fresh water. The complexes A and B were measured at 225 nm (ε = 4.25 × 104 M−1 cm−1) and 226 nm (ε = 2.38 × 104 M−1 cm−1), respectively, against the reagent blank using a Cary 300 BIO UV-Vis spectrophotometer (Varian, Palo Alto, CA).
000 rpm, 13000g, on a micro Centrifugette 4214) were removed for Pt complex quantification and replaced with fresh water. The complexes A and B were measured at 225 nm (ε = 4.25 × 104 M−1 cm−1) and 226 nm (ε = 2.38 × 104 M−1 cm−1), respectively, against the reagent blank using a Cary 300 BIO UV-Vis spectrophotometer (Varian, Palo Alto, CA).
In order to evaluate the physisorbed platinum complex with respect to the chemisorbed one, the apatite–platinum complex conjugates were washed twice using 1 mL of ultrapure water. The platinum complex in the washing water was quantified by ICP-OES analysis (for the Pt assay the selected analytical wavelength was 265 nm), and the residual drug (calculated as the difference between the total adsorbed drug and the drug removed by washing) was considered to be effectively bound to the apatite nanocrystals.
The isotherm data were fitted to different models in order to obtain comparable and physically interpretable parameters, which describe the adsorption process. The models of Langmuir,30 Freundlich,31 and Langmuir–Freundlich (also known as Sips)32 were tested for the isotherms. The curves were fitted to the experimental data using a nonlinear least-squares fitting algorithm to obtain the model parameters.
The generalized Langmuir model is shown in eqn (3) where Ce and Qe are the Pt-complex concentration in solution and the amount of Pt-complex adsorbed on apatite, respectively, Qm is the maximum saturation load, K is a constant often regarded as an affinity constant, and m and n are surface heterogeneity parameters.
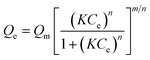 | (3) |
For the Langmuir model, the heterogeneity parameters m and n are both equal to 1 (eqn (4)), where KL is the Langmuir affinity constant.33 This basic model assumes an energetically homogeneous surface with the same adsorption energy for every adsorption site. The maximum adsorption capacity is a full monolayer coverage, and interaction between adsorbed species is not allowed.
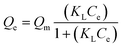 | (4) |
The Langmuir–Freundlich model takes into account the fact that the adsorption energy is not equal for all sites, as expressed by deviations of m and n from the value of 1. The model uses a Gaussian shaped energy distribution (m = n, eqn (5)) using a symmetric energy curve. The KLF is the affinity constant of the model.33
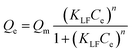 | (5) |
The Freundlich isotherm model (eqn (6), where KF is the Freundlich affinity constant), is an empirical model similar in form to the Langmuir–Freundlich model; however, it does not limit the adsorption to a monolayer and is often applicable to adsorption on heterogeneous surfaces.33
| Qe = KFC1/ne | (6) |
Determination of release kinetics
The solid apatite/complex conjugates were washed twice with ultrapure water and freeze-dried overnight at −60 °C under vacuum (3 mbar). An aliquot (about 5 mg) of the conjugate was mixed in a polyethylene tube with Hepes buffer saline solution (10 mM, 10 mL) containing chloride ions in the range 0.18–0.20 M. After 15 s of treatment in a vortex apparatus, the apatite suspension was maintained in a bascule bath at 37 °C. At scheduled times, aliquots (1 mL) of the supernatant (that was well separated from the solid phase by 6 min of centrifugation at 5000 rpm on a THERMO multi-speed PK 121) were removed for platinum and calcium ion quantification and replaced with fresh buffer. The supernatant was analyzed by UV-Vis spectroscopy and compared with the [PtCl2(en)] UV-vis spectrum. The amounts of platinum and calcium ions released from the HA nanocrystals were determined by ICP measurements as previously reported.Experiments with human cancer cells
Apatite/complex conjugate aliquots (about 10 mg) were mixed in a polyethylene tube with F12-HAM's medium (Sigma Chemical Co). After 15 s vortexing, the HA suspension was maintained in a bascule bath at 37 °C for 24 h. Subsequently, the supernatant (separated as described above) was removed and utilized for the cytotoxicity studies. Platinum released from the HA nanocrystals was determined by AAS-GF (graphite furnace atomic absorption) measurements. Platinum complexes A and B, as well as cisplatin, were dissolved in F12-HAM's medium just before the experiment.Cell cultures
Human lung (A549), cervical (HeLa), and colon (LoVo) carcinoma along with osteosarcoma (U2Os) cell lines were obtained by American Type Culture Collection (ATCC, Rockville, MD). Cell lines were maintained in the logarithmic phase at 37 °C in a 5% carbon dioxide atmosphere in Nutrient Mixture F-12 HAM's (Sigma Chemical Co.) containing 10% foetal bovine serum (Euroclone, Milan, Italy), antibiotics (50 units mL−1penicillin and 50 μg mL−1streptomycin), and 2 mM L-glutamine.Cytotoxicity assays
The cell growth inhibitory effect towards tumour cell lines was evaluated by means of an MTT assay.34 Briefly, 3–8 × 103cells per well, dependent upon the growth characteristics of the cell line, were seeded in 96-well microplates in growth medium (100 μL). After 24 h, the medium was removed and replaced with a fresh one containing the compound to be studied. Triplicate cultures were established for each treatment. After 72 h in the dark, each well was treated with 10 mL of 5 mg mL−1 MTT saline solution and, following 5 h of incubation, 100 μL of a sodium dodecylsulfate (SDS) solution in HCl 0.01 M were added. After an overnight incubation, the inhibition of cell growth induced by the tested complexes was detected by measuring the absorbance of each well at 570 nm using a Bio-Rad 680 microplate reader. Mean absorbance for each drug dose was expressed as a percentage of the control untreated well absorbance and plotted vs.drug concentration. IC50 values represent the drug concentrations that reduce the mean absorbance at 570 nm to 50% of those in the untreated control wells.AAS-GF analyses
Aliquots (1 mL) of the supernatant, obtained as described above and used for the experiments with human cells, were treated with 1 mL highly pure nitric acid (Pt ≤0.005 μg kg−1, TraceSELECT® Ultra, Sigma Chemical Co.) and transferred into a microwave Teflon vessel. Each mineralized sample was analyzed for Pt by using a Varian AA Duo graphite furnace atomic absorption spectrometer (Varian, Palo Alto, CA, USA) at 266 nm. The calibration curve was obtained using known concentrations of a standard Pt solution purchased from Sigma Chemical Co.Statistical analysis
Determinations of HA crystallite domain size, bulk and surface Ca/P ratio, ζ potential, and specific surface area were carried out 5 times on the same synthesis product. Data are presented as mean value ± standard deviation (SD). Adsorption, release, and cytotoxic experiments were performed in triplicate and results are reported as mean values ± standard deviation (SD). Data obtained from the experiments were compared by a two tailed t-test. Differences were considered statistically significant at a significance level of 90%.Results and discussion
Compositional and structural features of HA and CDHA
The codes, composition, specific surface area (SSA), and ζ potential of the two investigated materials are listed in Table 1. In both cases, the bulk Ca/P ratio obtained by ICP was lower than the stoichiometric one (1.67), but this latter was significantly lower for CDHA compared to HA due to the different amounts of Ca2+ and PO43− ions in the preparation procedures. For both materials, the surface Ca/P ratio determined by XPS was significantly lower than the bulk Ca/P ratio as a result of the nanocrystalline apatite surface disorder. A limited amount of carbonate, derived from CO2 dissolved in the preparation media and adsorbed on the surface materials during the storage, was present. The presence of carbonate in the structure of apatite was intentionally retained, in order to better mimick the biological ones.10 The carbonate amount was quantified according to the weight loss between 600 and 1000 °C35 (Table S1†). Both materials exhibited a high SSA in the 140–170 m2 g−1 range, which is in agreement with the nanometric size observed by TEM (see below). ζ potential measurements show that the CDHA has an enhanced negative charge compared to HA, which is in good agreement with its surface calcium deficiency.| Materials | Bulk Ca/Pa/mol | Surface Ca/Pb/mol | Carbonate speciesc (wt %) | SSABET/m2 g−1 | ζ Potential/mV | D 002 d/nm | D 310 d/nm | Degree of crystallinity |
|---|---|---|---|---|---|---|---|---|
| a Calculated by ICP-OES. b Calculated by XPS. c Calculated by TGA. d Calculated applying the Scherrer equation. | ||||||||
| HA | 1.62 | 1.45 | 1.3 | 160 ± 16 | −10.1 ± 0.8 | 22 ± 4 | 7 ± 3 | 61% ± 5 |
| CDHA | 1.50 | 1.20 | 2.2 | 150 ± 15 | −21.2 ± 1.4 | 23 ± 5 | 9 ± 3 | 65% ± 4 |
Powder X-ray diffraction patterns of HA and CDHA (Fig. 1) show the characteristic diffraction maxima of hydroxyapatite single phase (JCPDS 9-432). The diffraction patterns of HA, as well as of CDHA, exhibit not well defined diffraction maxima indicating a relatively low degree of crystallinity and nano-dimensions. The degrees of crystallinity of HA and CDHA, quantified according to a previously reported method, were 61 ± 5% and 65 ± 4%, respectively, and did not differ significantly. The average crystal sizes along the c-axis and along a perpendicular to it, were calculated by Scherrer's formula using the 2θ = 26° (002) and 2θ = 39° (310) diffraction peaks, respectively. As far as the degree of crystallinity is concerned, the crystallite domain sizes of both nanocrystals appeared to be very similar due to the same procedure and temperature used for their preparation. The values of the crystallite domain sizes are in good agreement with the dimensions calculated by TEM image (see below).
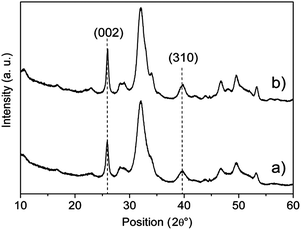 | ||
| Fig. 1 X-Ray diffraction patterns of HA (a) and CDHA (b). | ||
The FT-IR spectra of HA and CDHA (Fig. 2) confirm the presence of the typical adsorption bands of apatite. In agreement with the diffraction data, the low crystalline degree of both materials is supported by the weak resolution of all adsorption bands. Moreover, the better resolution of the apatite OH bands at 3570 and 632 cm−1 indicates an increase in hydroxylation of CDHA with respect to HA. This finding is in agreement with the enhanced surface negative charge of CHDA with respect to HA, as calculated by ζ potential measurements. The broad band around 3400 cm−1 and the band around 1640–1630 cm−1 are due to water adsorbed on the materials. The amount of adsorbed water is the same for both apatites in agreement with the weight losses between room temperature and 200 °C (Table S1†). The presence in the HA nanocrystals of a band at 1422 and of a broad band at 1460 cm−1 is consistent with a carbonate type-B substituted apatite, where the carbonate ions replace the phosphate ions in the crystal lattice.36 On the contrary, in the CDHA nanocrystals the presence of a band at 1454 cm−1 in addition to the previous two bands is consistent with type-B and type-A carbonate-substituted hydroxyapatite where the carbonate ions replace both the phosphate ions and the hydroxyl groups in the crystal lattice.36
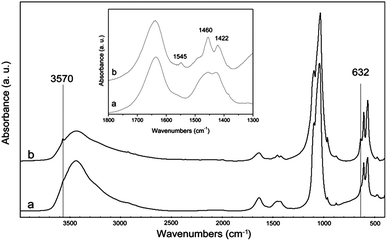 | ||
| Fig. 2 FT-IR spectra of HA (a) and CDHA (b). The inset shows the bands of carbonate ions in the region 1800–1300 cm−1. | ||
The chemical composition of biological apatite and, similarly, of nanocrystalline apatites, has been the object of several approximations (the Ca/P ratio in the bone is close to 1.5).10 A general chemical formula was proposed by Winand for HPO42− containing apatites (eqn (7)) and by Labarthe et al. for carbonate-containing apatites (eqn (8)).37
| Ca10−x(PO4)6−x(HPO4)x(OH)2−x with 0 ≤ x ≤ 2 | (7) |
| Ca10−x(PO4)6−x(CO3)x(OH)2−x with 0 ≤ x ≤ 2 | (8) |
These formulae establish a similar behaviour for substitution of bivalent carbonate for trivalent phosphate: that is the creation of a cationic vacancy (phosphate case) and of an anionic vacancy (carbonate case) in monovalent sites. These chemical formulae are consistent with the observed limit composition (x = 2) and with the decrease in the OH−apatite content when the amount of carbonate and/or HPO42− increases. Other chemical formulae have been proposed. The most general, which combines the presence of carbonate and HPO42−, is eqn (9):37
| Ca10−x(PO4)6−x(HPO4 or CO3)x(OH or ½CO3)2−x 0 ≤ x ≤ 2 | (9) |
Taking into account the latter formula, it is possible to hypothesize that the different amounts of HPO42− and CO32− are responsible for the different Ca/P ratios in HA and CDHA nanocrystals. When HPO42− ions are present, specific bands relative to this ion are observed in the FT-IR spectra. In fact, in most nanocrystalline apatite samples and especially in biological apatites, additional bands are observed which do not appear in well-crystallized apatites and which have been designated as “non-apatitic environments” of the mineral ions.37 The curve fitting of the bands characteristic of PO4 species indicates the presence, in both apatites, of two “non-apatitic” signals at 616 cm−1, due to PO43−, and at 533 cm−1, due to HPO42− (Fig. 3). These bands are slightly more intense for the CDHA, in agreement with its greater amount of phosphate ions in the lattice, responsible for the lower Ca/P ratio.
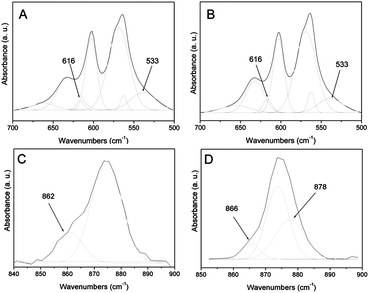 | ||
| Fig. 3 Curve fitting of the ν4PO4 bands of HA (A) and CDHA (B) and of ν3CO3 bands of HA (C) and CDHA (D). | ||
The study of the FT-IR bands characteristic of carbonate species ν2 (850–900 cm−1) and ν3 (1350–1600 cm−1) suggests type-B (carbonate substitution for phosphate) for HA and type-B as well as type-A (carbonate substitution for OH) for CDHA. The carbonate is incorporated in the apatite structure, with a very few carbonate ions on non-apatitic sites at the crystal surface. The curve fitting of the bands characteristic of carbonate species ν3 (Fig. 3) indicates the presence of a major component at 871 cm−1 due to carbonate type-B in both apatites, a band at 878 cm−1 assigned to carbonate type-A in the CDHA, and a band around 864 cm−1 corresponding to a labile carbonate environment in both apatites.38 The ratio “labile CO3/total CO3” was 0.3 for HA and 0.1 for CDHA. These values indicate that the CDHA has a higher tendency to incorporate carbonate in the lattice with respect to HA, due to its unbalanced charge, and that CO32− as well as HPO42− has an important role in the ionic balance of the CDHA, therefore in its Ca/P ratio.
TEM images representative of the morphological and structural features of CDHA and HA nanoparticles are displayed in Fig. 4A and B, respectively. Focussing on CDHA, the particle projections on the image plane exhibited typically three shapes: rectangles ca. 25 nm in length and 15 nm in width (e.g. particle A), ellipsoids ca. 15 × 8 nm (e.g. particle B), and rectangles ca. 25 nm in length and 8 nm in width (e.g., particle C). The two series of fringes present on particle B (see Fig. 4B, zoom), 8.10 Å apart and forming an angle of 120°, are due to (100) and (010) planes, perpendicular to the crystallographic a- and b-axis, respectively, of the HA structure. It can be inferred that an image-like that labelled B should result from the projections of a HA particle oriented with the c-axis perpendicular to the image plane. The elongation of projection with rectangular shapes (e.g. A and C) should correspond to particles oriented with the c-axis parallel to the image plane. In summary, it can be proposed that the morphology of CDHA particles is shaped as platelet with roundish borders, with dimensions (length × width × thickness) of ca. 25 × 15 × 8 nm.
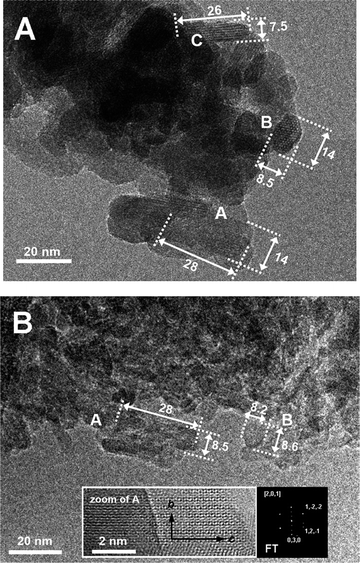 | ||
| Fig. 4 (A) TEM image representative of shape and size of CDHA particles. A, B, and C indicate three typical projection shapes. The units of the values reported near the arrows are nm. Original magnification: 150K× (B) TEM image representative of shape and size of HA particles. A and B indicate two typical projection shapes. The units of the values reported near the arrows are nm. Original magnifications: (i) image in the main frame = 150K×; zoomed image = 600K×. | ||
In the case of HA (Fig. 4B), similar particle images were observed (just slightly smaller in size). Unfortunately, it was not possible to clearly determine any zone axis for the circular/ellipsoidal projections. Conversely, the FT of systems of diffraction fringes associated to rectangular projections indicated that these latter should be produced by particles observed along the a-axis and oriented with the c-axis along the main dimension on the projection. Hence, it can be inferred that the HA sample is constituted of particles quite similar, in morphology and extension along the main crystallographic axes, to the CDHA sample, and this is in agreement with SSABET and XRD data.
It is worth noting that for both samples, the diffraction fringes associated to particle images extend in several cases up to the surface of the particles, whilst the borders appear quite irregular. This indicates that the particles are not terminated by specific crystalline planes.
Adsorption of platinum–bisphosphonate complexes onto apatite nanocrystals
Bis-{ethylenediamineplatinum(II)}-2-amino-1-hydroxyethane-1,1-diyl-bisphosphonate (A in Fig. 5) and bis-{ethylenediamineplatinum(II)}medronate (B in Fig. 5) were prepared as previously described.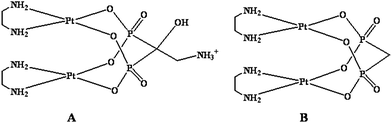 | ||
| Fig. 5 Sketches of platinum complexes used in this work. | ||
The adsorption isotherms of the two platinum complexes were investigated with respect to the HA and CDHA nanocrystals. In Fig. 6 the adsorbed amount of Pt complex, Γ (mg m−2), is plotted against its solution concentration after adsorption, CPt-complex (mg mL−1). The adsorption experiments have been performed at 37 °C in an aqueous medium over a period of 24 hours. The adsorbed amount of platinum complex was determined by the difference between the initial Pt complex and that remaining in the supernatant solution.
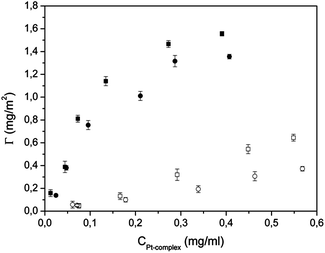 | ||
| Fig. 6 Adsorption isotherms of A onto HA (■) and CDHA (●) and of B onto HA (□) and CDHA (○). | ||
The profiles of the adsorption-isotherms for the two complexes towards the two types of apatites appear to be very different. The adsorption of A is characterized by a high initial slope, indicating a great Pt-complex affinity for the apatite surface. As the A concentration increases in the buffer solution, also the surface coverage increases, until completion. The adsorption saturation yields plateau values corresponding to a maximum amount of A immobilization on the surface of 1.6 and 1.4 mg m−2 for HA and CDHA, respectively. The shapes of the adsorption isotherms for complex A are very similar to a Langmuirian curve, indicating a high affinity for the apatite substrate. In contrast, the isotherms for adsorption of complex B cannot be described by the Langmuir model, or other similar models, since they appear almost linear, indicating scarce affinity for the apatites. Under these conditions, the isotherms do not reach plateau values and the maximum amount of B, in the experimental conditions used, was 0.67 and 0.31 mg m−2, for HA and CDHA, respectively. For each complex, the affinity is greater for HA than for CDHA and this appears to be related to the higher Ca/P ratio of HA. On the other hand, for each apatite, the affinity is greater for complex A than for complex B. Such a difference may be due to the positive charge (NH3+ moiety) of complex A which increases its attraction for the negatively charged apatites.
The Langmuir, Freundlich, and Langmuir–Freundlich models have been utilized to describe the adsorption isotherms of complex A. The corresponding curves, calculated with the best-fitting parameters, are displayed in Fig. S1†. In both cases the Langmuir–Freundlich model gives the best fit, which is also confirmed by the r2 values (Table 2). It is possible that the adsorption sites, along the apatite surface, are energetically heterogeneous, a factor which is taken into account in this model. The best-fitting adsorption parameters, calculated according to Langmuir, Freundlich, and Langmuir–Freundlich models, are reported in Table 2 for adsorption of A onto the two types of apatite nanocrystals. The surface heterogeneity of HA as well as of CDHA, evidenced at a molecular scale by the adsorption of Pt-complexes, is in agreement with the HRTEM observations at a nanometric scale, and with the typical surface characteristics of biomimetic apatites of low crystallinity and having a non-stoichiometric surface Ca/P molar ratio.39 This implies that the real adsorption systems could be a combination of two or more models. In the case of complex A, the constant KLF, which can be viewed as a measure of the affinity of the platinum–bisphosphonate complexes toward the apatitic surface, is significantly larger for HA than for CDHA. Such a trend suggests that the affinity of the bisphosphonate moiety for the apatitic calcium ions is more important than the electrostatic attraction between the positively charged –NH3+ residue (present on the bisphosphonate ligand of A) and the negatively charged apatite. The latter interaction would favour the attraction of A for the more negatively charged surface of CDHA.
| Models | Adsorption of A onto HA | Adsorption of A onto CDHA |
|---|---|---|
| Langmuir, KL/mL mg−1 | 7.2 ± 1.6 | 5.1 ± 1.4 |
| Langmuir, Qm/mg g−1 | 325.8 ± 30.5 | 322.0 ± 42.2 |
| Langmuir, r2 | 0.981 | 0.978 |
| Freundlich, KF/mL mg−1 | 406.5 ± 57.9 | 393.8 ± 53.8 |
| Freundlich, n | 1.9 ± 0.5 | 1.7 ± 0.3 |
| Freundlich, r2 | 0.937 | 0.947 |
| Langmuir–Freundlich, KLF/mL mg−1 | 11.9 ± 1.8 | 8.7 ± 1.9 |
| Langmuir–Freundlich, Qm/mg g−1 | 255.9 ± 20.3 | 257.6 ± 44.5 |
| Langmuir–Freundlich, n | 1.5 ± 0.3 | 1.3 ± 0.35 |
| Langmuir–Freundlich, r2 | 0.992 | 0.984 |
The adsorption over a time of 140 hours was also investigated. The adsorbed amount of platinum complex was determined by the difference between the initial Pt quantity and that remaining in the supernatant solution at a given time. Fig. 7 shows the adsorption profiles of the platinum complexes A and B on apatite nanocrystals, starting from a 1 mg mL−1 complex concentration in water. The plots show that the maximum uptake of complex A is reached after about 24 hours for both types of apatite nanocrystals. The final percentages of adsorbed complex A were about 100 and 97% for HA and CDHA, respectively. This corresponds to about 150 mg of complex per g of apatite. These values are in good agreement with those of the adsorption isotherms. In fact, after 24 hours most of the complex was adsorbed on the apatites.
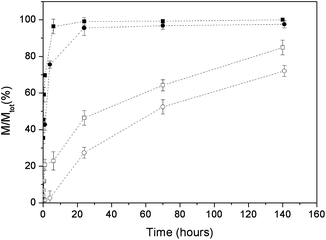 | ||
| Fig. 7 Adsorption of complex A onto HA (■) and CDHA (●) and of complex B onto HA (□) and CDHA (○). | ||
Fig. 7 also shows the adsorption profiles of complex B. The adsorption kinetics are similar for the two types of nanocrystals. After 140 h contact, the percentages of adsorbed complex were 84 and 72% for HA and CDHA, respectively. This corresponds to about 126 and 108 mg of complex B per g of HA and CDHA, respectively. In the case of B, the total amount of adsorbed complex is significantly smaller for CDHA than for HA, this was not the case for complex A. It is possible that in the case of complex A the lower affinity of the phosphonate for CDHA, expected on the basis of its lower calcium content, is compensated by a greater attraction between the positively charged residue present on the bisphosphonate ligand (–NH3+) and the more negatively charged surface of CDHA.
Complex A has a greater affinity for both types of nanoparticles, not only in terms of the total uptake but also in terms of the adsorption rate. For both types of nanocrystals, the uptake of complex A reaches its maximum after about 24 hours. In contrast, the uptake of complex B after 24 hours is significantly lower than that reached after 140 hours exposure. It is also worth noting that, while complex A reaches a plateau, being almost totally absorbed after 20 hours, in contrast complex B never reaches a steady state during the experimental period.
Two water washings were performed in order to distinguish between the physisorbed and the chemisorbed platinum complex. Moreover, the drug release experiments reported in the section below were carried out only upon the chemisorbed complex.
The chemisorbed complex A (mg per g of apatite) was 120 and 105 for HA and CDHA, respectively, while the chemisorbed complex B was 90 and 70 for HA and CDHA, respectively. For both complexes the loading is greater towards HA, which is characterized by a higher Ca/P ratio, than towards CDHA. It is to be noted that the differences between the two apatites become more evident by comparing the chemisorbed complex rather than the total (chemisorbed + physisorbed) complex.
Release of platinum–bisphosphonate complexes from apatite nanocrystals
The release experiments were performed at 37 °C in aqueous Hepes buffer (NaCl concentration in the range 0.18–0.20 mM) and monitored for a period of 7 days. The release profiles of complexes A and B from apatite nanocrystals are plotted in Fig. 8. The data are expressed in terms of M/Mtot, where Mtot is the chemisorbed complex (the amount of complex effectively bound to the HA nanocrystals).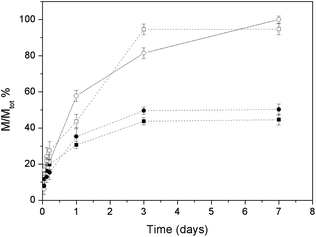 | ||
| Fig. 8 Released mass percentages of complex A from HA (■) and CDHA (●) and of complex B from HA (□) and CDHA (○). | ||
The release profiles are quite similar for the two types of apatite nanocrystals, but depend very much upon the type of complex. The total amounts of complex released after 7 days exposure are about 50% for complex A (44 and 50% of the chemisorbed quantities for HA and CDHA, respectively) and about 100% for complex B (100 and 94% of the chemisorbed quantities for HA and CDHA, respectively). The rate of release is faster for complex B (for which the percentage of release is greater) than for complex A (for which the percentage of release is smaller).
The smaller release of chemisorbed complex in the case of A deserves a comment. As will be shown in the next section, in all cases platinum is released as a Pt(en) chlorido or solvato species while the bisphosphonate remains anchored to the apatite matrix. In the case of A the BP contains a free aminic function that can coordinate a metallic residue impeding its release to the solution.
Characterization of the released Pt species
In order to characterize the Pt species released from the two different types of apatite nanocrystals, UV-Vis experiments have been performed to compare the spectra of complexes A and B with the platinum precursor [PtCl2(en)] (Fig. S2†). The UV-Vis spectrum of the released platinum species is similar to that of [PtCl2(en)], indicating that the complex released is [PtCl2(en)] (or related solvato species) in agreement with results found in previous work.26Calcium release from apatite nanocrystals before and after loading with platinum–bisphosphonate complexes
The amounts of calcium ions released in Hepes buffer saline solution from HA and CDHA nanocrystals, as a function of time, are plotted in Fig. 9. After 7 days the calcium released was 3.4 and 2.9% of the total content for HA and CDHA, respectively. This result is coherent with the higher amount of calcium present in the HA with respect to the CDHA nanocrystals.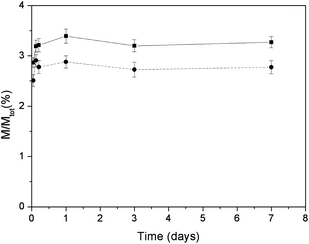 | ||
| Fig. 9 Released mass percentages of calcium ions as a function of time for HA (■) and CDHA (●). | ||
The mass percentages of released calcium ions, as a function of time, for the two apatites loaded with complexes A and B are reported in Fig. 10. In the case of complex A, the amounts of calcium released after 7 days exposure were 4.0 and 3.7% of the total calcium content for HA and CDHA, respectively. Both values are slightly greater than the calcium released from untreated apatites. In the case of complex B, the amounts of calcium ions released from the loaded HA and CDHA nanocrystals were 3.3 and 3.0% of the total calcium content, respectively. In this latter case, the released amounts of calcium ions are quite similar to that released from untreated apatites. In the case of complex A the adsorption by HA and CDHA was in accord with a Langmuirian profile, which implies an irreversible adsorption. Therefore, the adsorption of complex A could increase the release of calcium ions from the apatites. In contrast, the linear adsorption profile observed for complex B implies a reversible adsorption which can take place without release of calcium. In all cases, the amount of released calcium is rather small in agreement with the apatite low solubility at neutral pH.
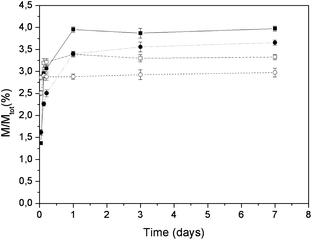 | ||
| Fig. 10 Released mass percentages of calcium ions as a function of time for loading of complex A on HA (■) and CDHA (●) and for loading of complex B on HA (□) and CDHA (○). | ||
Cytotoxicity assay and AAS-GF analyses
Unmodified and Pt-adsorbed HA nanocrystals were both evaluated for their cytotoxic properties against human cervical (HeLa), colon (LoVo), and lung (A549) cancer cells as well as against osteosarcoma (U2Os) cells. For comparison purposes, the cytotoxicity of A and B was evaluated at the same Pt concentration of that released in F12 Ham's media from Pt-adsorbed HA nanocrystal. Pt concentrations and cell viability after 72 h drug exposure are summarized in Table 3.| Compound | Pt/μM | Cell viability (%) ± SD | |||
|---|---|---|---|---|---|
| HeLa | LoVo | U2Os | A549 | ||
| a SD = standard deviation. | |||||
| CDHA | 0 | 96.24 ± 1.24 | 97.41 ± 3.08 | 98.13 ± 3.44 | 97.70 ± 3.32 |
| A | 13.77 | 69.32 ± 2.05 | 76.43 ± 2.41 | 56.43 ± 1.04 | 78.54 ± 2.26 |
| A loaded CDHA | 13.77 | 46.25 ± 3.05 | 62.48 ± 2.92 | 51.01 ± 2.42 | 72.24 ± 2.26 |
| [PtCl2(en)] | 13.77 | 44.23 ± 2.26 | 61.27 ± 2.06 | 49.09 ± 2.15 | 68.54 ± 3.28 |
| B | 17.56 | 59.27 ± 2.11 | 71.11 ± 2.26 | 67.43 ± 2.63 | 77.27 ± 2.17 |
| B loaded CDHA | 17.56 | 40.43 ± 2.31 | 57.43 ± 2.33 | 53.43 ± 3.11 | 69.15 ± 1.26 |
| [PtCl2(en)] | 17.56 | 40.30 ± 3.21 | 54.24 ± 2.76 | 51.07 ± 3.48 | 65.51 ± 2.48 |
| HA | 0 | 92.47 ± 1.65 | 93.46 ± 1.78 | 90.37 ± 1.08 | 94.61 ± 1.20 |
| A | 5.61 | 75.23 ± 1.52 | 88.54 ± 3.16 | 82.43 ± 2.25 | 91.61 ± 2.35 |
| A loaded HA | 5.61 | 60.04 ± 2.43 | 81.37 ± 1.98 | 73.34 ± 1.87 | 82.43 ± 1.76 |
| [PtCl2(en)] | 5.61 | 62.23 ± 2.13 | 80.56 ± 3.24 | 71.01 ± 2.82 | 79.20 ± 2.65 |
| B | 7.38 | 71.96 ± 2.23 | 82.24 ± 2.13 | 77.65 ± 2.24 | 88.27 ± 2.43 |
| B loaded HA | 7.38 | 56.03 ± 1.97 | 75.23 ± 1.19 | 68.13 ± 1.85 | 75.65 ± 2.25 |
| [PtCl2(en)] | 7.38 | 56.41 ± 2.06 | 73.32 ± 2.53 | 65.23 ± 2.06 | 73.24 ± 2.98 |
Unmodified HA and CDHA nanocrystals proved to be completely ineffective in decreasing cell viability of all tumor cell lines. Free A complex determined an in vitro antitumor activity that was roughly 1.2 and 1.4 fold lower than that obtained for A complex loaded on HA and CDHA nanocrystals, respectively. Therefore, loading on CDHA nanocrystals increased the cytotoxicity of A by about 55% in HeLa and U2Os cells, and by roughly 30% in LoVo and A549 cells. On the other hand, loading on HA nanocrystals increased the cytotoxicity of A by the same amount (24%) towards all tested cell lines. Similarly to complex A, also complex B antiproliferative activity was increased by 1.2 and 1.3-times after loading on HA and CDHA nanocrystals, respectively. In particular, loading of B on CDHA and HA nanocrystals determines a decrease in HeLa cell viability by about 60 and 45%, respectively. Interestingly, the cytotoxicity of A and B loaded on nanocrystals is similar to that of free [PtCl2(en)]. These results support the hypothesis that the species released from A and B loaded on nanocrystals is [PtCl2(en)] (or related solvato species).
Conclusions
The aim of this study was to test nanocrystalline apatites as bone-specific drug delivery devices to be used for the treatment of bone tumours either by local implantation or by injection. In particular, we were interested in unraveling how the Ca/P ratio can influence the upload and release of anticancer platinum–bisphosphonate complexes. To this end, two types of nanosized and poorly crystalline biomimetic apatites having similar chemico-physical properties in terms of morphology, degree of crystallinity, dimensions, and surface area but different Ca/P ratio (apatite, HA, and calcium deficient apatite, CDHA) were synthesized and fully characterized. In previous works it was found that the Ca2+ ions act as anchoring sites for the bisphosphonate ligand and influence the nature of the platinum complex released in solution.25,27The adsorption isotherms and adsorption kinetics of two platinum complexes with different bisphosphonate ligands, A and B, onto the synthesized nanocrystals have been investigated. Both complexes contain a geminal bisphosphonate but complex A contains, in addition, a charged ammonium group. This structural difference dramatically increases the affinity of A for the apatite nanocrystals, in agreement with previous work.26
The isotherm–adsorption profiles appear to be very different for the two complexes. For complex A the shape of the adsorption isotherms is very similar to a Langmuirian curve, indicating a high affinity for the apatite substrate. A comparison among Langmuir, Freundlich, and Langmuir–Freundlich models indicates that for both apatites (HA and CDHA) the Langmuir–Freundlich model gives the best fit. However, the constant KLF, which can be viewed as a measure of the affinity of the platinum–bisphosphonate complex toward the apatite surface, is significantly larger for HA than for CDHA. Unlike the case of complex A, the isotherms of complex B cannot be described by the Langmuir model, or other similar models, since these isotherms are almost linear indicating a scarce affinity for apatites. Therefore, the isotherms do not reach plateau values and, in the experimental conditions used, the amount of adsorbed B is higher for HA than for CDHA. The greater affinity for HA of both complexes appears to be related to its higher Ca/P ratio. On the other hand, the greater affinity of complex A for both apatites may be due to its positive charge (–NH3+ moiety) which increases its attraction for the negatively charged apatites.
The adsorption kinetics confirm the findings previously described for the adsorption isotherms. In fact, the comparison between the two platinum complexes clearly shows that complex A has a greater affinity for both types of nanoparticles, not only in terms of the total uptake but also in terms of the adsorption rate.
The release of the platinum complexes from the apatite nanoparticles follows a trend opposite to that observed for the adsorption process. Complex A, which was the most effective in the adsorption process, is the less effective in the desorption. This could be due to the aminic group present on the BP of complex A which remains anchored to the apatite matrix and can coordinate to platinum impeding its release to the solution.
The calcium released from complex A-loaded apatites is generally greater than that released from untreated apatites. This finding is in agreement with an irreversible adsorption of complex A on both apatites (Langmuirian profile), and the release of calcium from the apatites is probably connected to an erosion of the matrix. In contrast, the calcium released from complex B-loaded apatites is similar to that released from untreated apatites. This is in accord with the linear adsorption profile of complex B which implies reversible adsorption without alteration of the matrix. In all cases, the amount of calcium released is rather small, in agreement with the apatite low solubility at neutral pH.
The UV-Vis analysis showed that the released species is the same for complexes A and B and matches the behaviour of [PtCl2(en)] under similar conditions, thus indicating that the platinum–bisphosphonate bonds are broken in the release process.26Apatite-adsorbed and free Pt complexes were tested for their cytotoxicity towards human cervical (HeLa), colon (LoVo), and lung (A549) cancer cells as well as against osteosarcoma (U2Os) cells. The apatite-loaded Pt complexes were more cytotoxic than the unmodified compounds A and B and their cytotoxicity was comparable to that of [PtCl2(en)], thus indicating a common active species.
The above results demonstrate that apatite nanocrystals and antitumor drugs can be conjugated in such a way to yield a smart bone filler delivery system, acting both as a bone substitute and as a platinum drug releasing agent with the final goal of locally inhibiting the tumor re-growth and reducing the systemic toxicity. The materials here described not only can ensure a prolonged release of active species but also can improve the performance of the unmodified drug. Moreover, these results confirm the possibility of using the chemico-physical differences of apatite nanocrystals, in this case the Ca/P ratio, in order to tailor the release kinetics of the platinum complex.
Acknowledgements
We thank the Universities of Bari, Bologna, Piemonte Orientale, Padova, and Torino (funds for selected research topics), the Italian Ministero dell'Istruzione, Università e Ricerca (MIUR), the research project “Nanoparticelle multifunzionali per la terapia dei carcinomi: studi in vitro e in vivo in modelli sperimentali” funded by Regione Piemonte (Progetto Ricerca Sanitaria Finalizzata 2009, Grant no: 30258/DB2001) and the Inter-University Consortium for Research on Chemistry of Metals in Biological Systems (C.I.R.C.M.S.B). M.I. is recipient of a fellowship from Regione Piemonte. Dr Luca Bertinetti (ex University of Torino) is kindly acknowledged for TEM images of CDHA.Notes and references
- S. Pietronave, M. Iafisco, D. Locarno, L. Rimondini and M. Maria Prat, J. Appl. Biomater. Biomech., 2009, 7, 77–89 CAS.
- D. Peer, J. M. Karp, S. Hong, O. C. FaroKHzad, R. Margalit and R. Langer, Nat. Nanotechnol., 2007, 2, 751–760 CrossRef CAS.
- K. J. Cho, X. Wang, S. M. Nie, Z. Chen and D. M. Shin, Clin. Cancer Res., 2008, 14, 1310–1316 CrossRef CAS.
- H. Hillaireau and P. Couvreur, Cell. Mol. Life Sci., 2009, 66, 2873–2896 CrossRef CAS.
- J. H. Adair, M. P. Parette, E. I. Altınoğlu and M. Kester, ACS Nano, 2010, 4, 4967–4970 CrossRef CAS.
- L. M. Bareford and P. W. Swaan, Adv. Drug Delivery Rev., 2007, 59, 748–758 CrossRef CAS.
- G. Russell-Jones, K. McTavish, J. McEwan, J. Rice and D. Nowotnik, J. Inorg. Biochem., 2004, 98, 1625–1633 CrossRef CAS.
- L. Bildstein, C. Dubernet and P. Couvreur, Adv. Drug Delivery Rev., 2011, 63, 3–23 CrossRef CAS.
- S. E. Leucuta, Curr. Clin. Pharmacol., 2010, 5, 257–280 CrossRef CAS.
- N. Roveri, B. Palazzo and M. Iafisco, Expert Opin. Drug Delivery, 2008, 5, 861–877 CrossRef CAS.
- M. Epple, K. Ganesan, R. Heumann, J. Klesing, A. Kovtun, S. Neumann and V. Sokolova, J. Mater. Chem., 2010, 20, 18–23 RSC.
- V. Sokolova and M. Epple, Nanoscale, 2011, 3, 1957–1962 RSC.
- V. Uskokovic and D. P. Uskokovic, J. Biomed. Mater. Res., Part B, 2011, 96, 152–191 CrossRef.
- M. Iafisco, M. D. Foggia, S. Bonora, M. Prat and N. Roveri, Dalton Trans., 2011, 40, 820–827 RSC.
- M. Zhang and K. Kataoka, Nano Today, 2009, 4, 508–517 CrossRef CAS.
- M. Stigter, J. Bezemer, K. de Groot and P. Layrolle, J. Controlled Release, 2004, 99, 127–137 CrossRef CAS.
- F. Errassif, A. Menbaoui, H. Autefage, L. Benaziz, S. Ouizat, V. Santran, S. Sarda, A. Lebugle, C. Combes, A. Barroug, H. Sfihi and C. Rey, in Advances in Bioceramics and Biotechnologies: Ceramic Transactions, ed. R. Narayan, M. Singh and J. McKittrick, Wiley-VCH Verlag GmbH & Co. KGaA, 2010, vol. 218 Search PubMed.
- P. J. O'Dwyer, J. P. Stevenson and S. W. Johnson, in Cisplatin: Chemistry and Biochemistry of a Leading Anticancer Drug, ed. B. Lippert, Verlag Helvetica Chimica Acta. Zurich, 1999, pp. 29–69 Search PubMed.
- G. Daugaard and U. Abildgaard, Cancer Chemother. Pharmacol., 1989, 25, 1–9 CrossRef CAS.
- E. Wong and C. M. Giandomenico, Chem. Rev., 1999, 99, 2451–2466 CrossRef CAS.
- S. Zhang, G. Gangal and H. Uludag, Chem. Soc. Rev., 2007, 36, 507–531 RSC.
- S. Josse, C. Faucheux, A. Soueidan, G. Grimandi, D. Massiot, B. Alonso, P. Janvier, S. Laïb, P. Pilet, O. Gauthier, G. Daculsi, J. Guicheux, B. Bujoli and J.-M. Bouler, Biomaterials, 2005, 26, 2073–2080 CrossRef CAS.
- G. H. Nancollas, R. Tang, R. J. Phipps, Z. Henneman, S. Gulde, W. Wu, A. Mangood, R. G. G. Russell and F. H. Ebetino, Bone, 2006, 38, 617–627 CrossRef CAS.
- R. G. Russell, N. B. Watts, F. H. Ebetino and M. J. Rogers, Osteoporosis Int., 2008, 19, 733–759 CrossRef CAS : a journal established as result of cooperation between the European Foundation for Osteoporosis and the National Osteoporosis Foundation of the USA.
- B. Palazzo, M. Iafisco, M. Laforgia, N. Margiotta, G. Natile, C. L. Bianchi, D. Walsh, S. Mann and N. Roveri, Adv. Funct. Mater., 2007, 17, 2180–2188 CrossRef CAS.
- M. Iafisco, B. Palazzo, M. Marchetti, N. Margiotta, R. Ostuni, G. Natile, M. Morpurgo, V. Gandin, C. Marzano and N. Roveri, J. Mater. Chem., 2009, 19, 8385–8392 RSC.
- N. Margiotta, R. Ostuni, D. Teoli, M. Morpurgo, N. Realdon, B. Palazzo and G. Natile, Dalton Trans., 2007, 3131–3139 RSC.
- S.-C. Liou, S.-Y. Chen, H.-Y. Lee and J.-S. Bow, Biomaterials, 2004, 25, 189–196 CrossRef CAS.
- L. Bertinetti, A. Tampieri, E. Landi, C. Ducati, P. A. Midgley, S. Coluccia and G. Martra, J. Phys. Chem. C, 2007, 111, 4027–4035 CAS.
- I. Langmuir, J. Am. Chem. Soc., 1918, 40, 1361–1403 CrossRef CAS.
- H. M. F. Freundlich, Z. Phys. Chem., Stoechiom. Verwandtschaftsl., 1906, 385–471 CAS.
- R. Sips, J. Chem. Phys., 1948, 16, 490–495 CrossRef CAS.
- C. Rill, Z. I. Kolar, G. Kickelbick, H. T. Wolterbeek and J. A. Peters, Langmuir, 2009, 25, 2294–2301 CrossRef CAS.
- M. C. Alley, D. A. Scudiero, A. Monks, M. L. M. J. Hursey Czerwinski, D. L. Fine, B. J. Abbott, J. G. Mayo, R. H. Shoemaker and M. R. Boyd, Cancer Res., 1988, 48, 589–601 CAS.
- M. Iafisco, E. Varoni, E. Battistella, S. Pietronave, M. Prat, N. Roveri and L. Rimondini, Int. J. Artif. Organs, 2010, 33, 765–774 CAS.
- M. Iafisco, J. G. Morales, M. A. Hernandez-Hernandez, J. M. Garcia-Ruiz and N. Roveri, Adv. Eng. Mater., 2010, 12, B218–B223 CrossRef.
- C. Rey, C. Combes, C. Drouet, H. Sfihi and A. Barroug, Mater. Sci. Eng., C, 2007, 27, 198–205 CrossRef CAS.
- C. Rey, B. Collins, T. Goehl, I. R. Dickson and M. J. Glimcher, Calcif. Tissue Int., 1989, 45, 157–164 CrossRef CAS.
- C. Jäger, T. Welzel, W. Meyer-Zaika and M. Epple, Magn. Reson. Chem., 2006, 44, 573–580 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: (1) Thermal stabilities of HA and CDHA. (2) Curve fitting of the adsorption isotherms of complex A on HA and on CDHA according to the Langmuir, Langmuir–Freundlich, and Freundlich models. (3) UV-Vis spectra of Pt-complexes, [PtCl2(en)], and the species released from apatite nanocrystals loaded with the platinum–bisphosphonate complexes. See DOI: 10.1039/c1nr11147g |
| This journal is © The Royal Society of Chemistry 2012 |