Reorganization of perylene bisimide J-aggregates: from delocalized collective to localized individual excitations†
Yuxi
Tian
a,
Vladimir
Stepanenko
b,
Theo E.
Kaiser
b,
Frank
Würthner
*b and
Ivan G.
Scheblykin
*a
aChemical Physics, Lund University, Box 124, 22100, Lund, Sweden. E-mail: ivan.scheblykin@chemphys.lu.se; Fax: +46 46 22 24119; Tel: +46 46 22 24848
bUniversität Würzburg, Institut für Organische Chemie and Röntgen Research Center for Complex Material Systems, Am Hubland, 97074, Würzburg, Germany. E-mail: wuerthner@chemie.uni-wuerzburg.de; Fax: +49 931 31 84756; Tel: +49 931 31 85340
First published on 10th November 2011
Abstract
Water-induced reorganization of individual one-dimensional J-aggregates of perylene bisimide (PBI) dyes was observed by fluorescence microscopy. Fluorescence spectra and decay kinetics of individual J-aggregates immobilized on glass surfaces were measured under a dry nitrogen atmosphere and under humid conditions. The fluorescence properties of PBI J-aggregates arisen from collective excitons under dry nitrogen atmosphere were changed to those of non-interacting dye monomers when water vapor was introduced into the environment (sample chamber). Time-dependent changes of the fluorescence spectra and lifetimes upon exposure to water vapor suggest an initial coordination of water molecules at defect sites leading to the formation of H-type dimer units that act as exciton quenchers, and a subsequent slower disintegration of the hydrogen-bonded J-aggregate into monomers that lack resonance coupling. Our present studies resulted in a direct demonstration of how drastically the optical properties of molecular ensembles and characteristics of their excited states can be changed by delicate reorganization of dye molecules at nanometre scales.
Introduction
Discovered by Jelley and Scheibe in the 1930s,1,2 J-aggregates have attracted more and more interest due to their potential applications as light-harvesting and energy-transporting media.3–5 Notably, in natural light-harvesting (LH) systems, such as LH II complexes and chlorosomes of photosynthetic bacteria, chlorophylls are arranged as J-aggregates making light-harvesting very efficient.6,7 Red-shifted, narrow-band absorption and fluorescence spectra are valuable characteristic features of J-aggregates. Strong coupling between monomers and exciton delocalization over at least several monomers at room temperature and up to 100 monomers at cryogenic temperatures are probable reasons for the excellent exciton transport properties in such materials.5,8 Moreover, exciton migration over millions of dye monomers has been reported for J-aggregates at low temperatures.9,10Perylene bisimide (PBI) dyes11 possess outstanding optical and electronic properties and they are thermally and photochemically highly stable. Thus these dyes have been used as electronic materials in field-effect transistors,12,13 electrophotographic14 and photovoltaic devices.15,16 Due to strong π–π interactions, PBI monomers readily form different types of aggregates even in organic solvents.11 The π–π stacking energy was found to be dependent on the substituents at the bay area (1, 6, 7, 12-positions) of the PBI core and imide nitrogens as well as on solvents. Organization of monomers in PBI aggregates can be controlled by modification of substituents and proper choice of the solvent, which determine the balance between the π–π interaction and other noncovalent forces (e.g., hydrogen bonding).17 Thus the structural organization of such aggregates is very sensitive to the environment. Recently it has been reported that PBI derivatives containing hydrogen atoms at imide nitrogens and solubilizing trialkoxyphenyl substituents at bay positions self-assemble into J-aggregates in non-polar solvents by hydrogen bonding and π–π interaction to give extended double string cables (Fig. 1).18,19
 | ||
| Fig. 1 Chemical structure of hydrogen bonding PBIs and illustration of their J-type aggregation. | ||
Since the pioneering work of Moerner and Oritt at the end of 1980s,20,21 single molecule spectroscopy (SMS) has become one of the most popular techniques over the last few years to study optical properties of individual molecules or nano-objects including J-aggregates without ensemble averaging inherent in bulk samples.22 The high fluorescence quantum yield and stability of PBI J-aggregates provide the opportunity to study them at the single aggregate level. Recently, efficient exciton migration along individual PBI J-aggregate chains was reported on the basis of fluorescence brightness measurements.23 In the present contribution, we report on the dramatic change of exciton coupling in such hydrogen bonding PBI derivative (PBI 1, Fig. 1) upon exposure to moisture as revealed by fluorescence microscopy measurements.
Experimental section
Sample preparation
The hydrogen bonding PBI 1 was synthesized according to a previously reported method.18,19 Spectrophotometric grade methylcyclohexane (MCH) was purchased from Sigma-Aldrich and used as received. Samples for fluorescence microscopy were prepared by spin-casting 10−7 to 10−8 M solutions of PBI 1 in MCH on microscope glass cover slips (Menzel-Glaser). Note that J-aggregates in such diluted solutions are thermodynamically unstable and that dissociation to monomers occurs within 1–2 h at room temperature. Therefore, the time period between the dilution of the thermodynamically stable concentrated stock solution (6 × 10−5 M) and spin-casting can be used to adjust the length of the aggregates in the resulting samples. In this work, we spin-cast the solution immediately after the dilution to obtain long aggregates. The cover slips were used either directly (referred to as “untreated glass”) or after rinsing by deionized water and dried under N2 flow (referred to as “treated glass”).SMS measurements
For the SMS studies, a home-built wide-field fluorescence microscope setup based on an Olympus IX-71 inverted microscope and a Princeton Instruments PhotonMAX 512 EMG CCD camera with on-chip multiplication gain was used.24 The aggregates and PBI 1 monomers were excited at 514 nm by using a CW Ar ion laser. The fluorescence was collected by an oil immersion objective lens (Olympus, UPlanFLN, 60×, NA = 1.25, Japan) and imaged on the CCD camera. Nitrogen gas was used during the measurements to reduce photobleaching. For the measurement of fluorescence spectra, a transmission holographic diffraction grating (150 grooves mm−1, Thorlabs) was placed in front of the CCD camera. The spectral sensitivity was calibrated by using a standard tungsten incandescent lamp and all spectra were corrected accordingly. The instrumental response function had a spectral full width at half-maximum (FWHM) of ∼6 nm.Lifetime measurements
For the time-resolved measurements, we have used the setup described previously.24 Briefly, in the fluorescence microscope the light of the second harmonic (spectral maximum at 458 nm) of a 150 fs pulse from a Ti-Sapphire laser (Tsunami, 80 MHz repetition rate) with spectral maximum at 916 nm was used. The fluorescence was detected by a fast avalanche photodiode (APD, Micro Photon Devices, crystal size 100 μm, 250 cps dark counts) and counted by a PicoHarp 300 (PicoQuant GmbH) photon counting system. Time-tagged time-resolved (TTTR) mode was used in order to obtain fluorescence decay kinetics and fluorescence intensity at any given time.Moisture treatment
The moisture treatment of samples was achieved by saturating the N2 gas flow used for the sample protection with water vapor. The N2 was pumped through hot water (55 °C) before reaching the sample chamber. The distance between the bottle with the hot water and the sample was long enough to let the nitrogen gas cool down to room temperature prior to reaching the sample chamber.Atomic force microscopy (AFM)
AFM measurements were performed under ambient conditions using a Veeco Multimode Nanoscope IV system operating in tapping mode in air. The AFM images were recorded by using the E-scanner with a maximum scan area of 15 × 15 μm. Silica cantilevers (OMCL-AC160TS, Olympus) with a resonance frequency of ∼300 kHz and a nominal spring constant of 42 Nm−1 were used. The freshly prepared dilute solution of J-aggregates in MCH with a concentration of c = 5 × 10−8 M was spin-coated onto glass substrates under 1100 rpm.Results
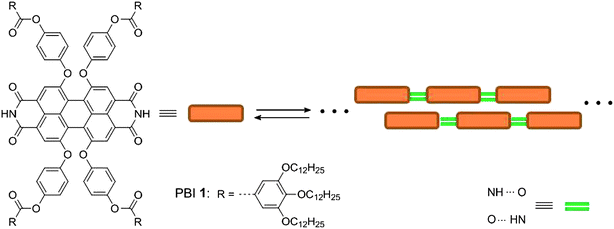
AFM images of PBI 1 aggregates spin-coated on untreated and treated glass substrates revealed similar thread-like aggregate shape (Fig. 2c and f). This shape of individual aggregates longer than 1 micrometre can also be resolved on the fluorescence microscopy images (Fig. 2a and d). Despite their similar shape, the fluorescence intensities and spectra of aggregates on the untreated and treated glasses are significantly different (Fig. 2b and e). On untreated glass substrate the fluorescence spectrum is quite similar to that of the J-aggregates in solution, with a maximum at ∼658 nm.18 Whereas on treated glass substrate the fluorescence intensity is much lower and the spectrum is broader and blue-shifted with a maximum at ∼605 nm, resembling rather the spectrum of the PBI 1 monomers in solution.18 For PBI monomers, the absorption cross-section is ∼2 times higher than that of the J-aggregates at the excitation wavelength of 514 nm. Surprisingly the fluorescence intensity is significantly lower on the treated surface indicating strong fluorescence quenching.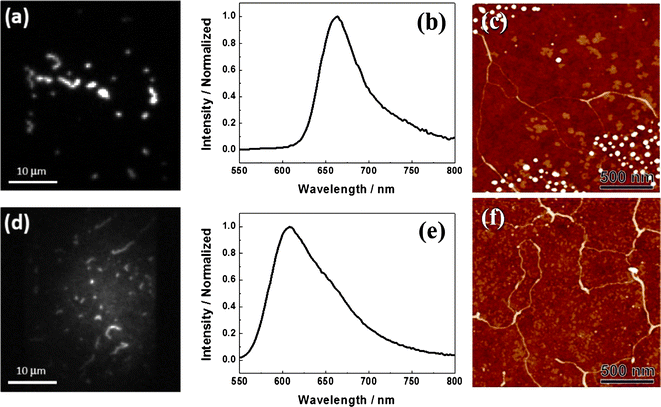 | ||
| Fig. 2 Fluorescence images (a and d) and spectra (b and e), and AFM images (c and f) of individual PBI 1 aggregates adsorbed on untreated (a, b and c), and treated (d, e and f) glass substrates. | ||
Fluorescence spectra of an aggregate on the untreated glass measured during the water vapor treatment are shown in Fig. 3. Initially, a pure J-aggregate spectrum was observed with a maximum at ∼658 nm, which is similar to the spectrum shown in Fig. 2b. When water vapor was conducted into the sample chamber, the fluorescence intensity of the aggregate decreased rapidly without any spectral shift (Fig. 3, inset (a)), and the emission was almost erased after about 80 s. After this time a broader and blue-shifted emission arose with a gradual increase of the intensity up to about 500 s showing a maximum at ∼605 nm (Fig. 3, inset (b)), which is identical to the spectrum shown in Fig. 2e and similar in shape to the spectrum of PBI 1 monomers on treated glass (Fig. S1 in the ESI†), indicating that the fluorescence observed after water vapor treatment originates from non-interacting PBI 1 monomers. Note that the spectrum of monomers on the treated glass is red-shifted by 20 nm compared to that on the untreated glass (Fig. S1 in the ESI†). This spectral shift most probably reflects the difference of the dielectric properties of the local environments of isolated dye molecules on untreated and treated glasses. As reported previously, the emission spectra of PBI 1 monomers are different in different solvents.19 The presence of water can change local environment of individual aggregates which may lead to a spectral shift.
 | ||
| Fig. 3 Time-dependent change of fluorescence intensity (λex = 514 nm) and fluorescence spectra (insets) of an individual thread-like aggregate chain of PBI 1 on untreated glass during the water vapor treatment (the treatment was started at time zero). | ||
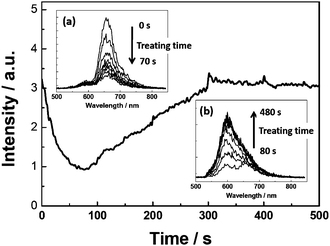
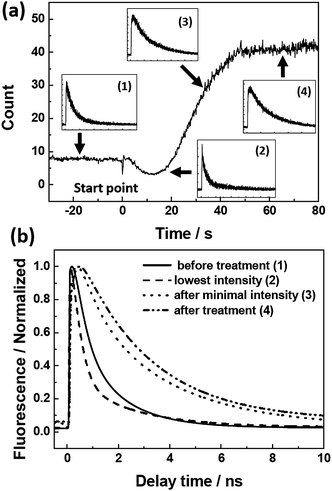
Fluorescence decays were also measured for a single aggregate during the vapor treatment (Fig. 4). The insets (1)–(4) in Fig. 4 show the fluorescence kinetics at the corresponding time points of the treatment as indicated. Fitting curves to the normalized decays are also presented for comparison (Fig. 4b). The fluorescence lifetime after the vapor treatment (Fig. 4a, inset (4)) obtained from the mono-exponential fit is ∼3.0 ns25 which corresponds to the fluorescence lifetime of PBI 1 monomers on the treated glass. However, bi-exponential functions had to be used for fitting the decay curves at other time points of the vapor treatment. The two components of the lifetime are 1.5 ns and 0.15 ns25 at the start point (Fig. 4a, inset (1)).
 | ||
| Fig. 4 (a) Fluorescence intensity trace of an individual J-aggregate chain on untreated glass during the water vapor treatment (0–330 s). Insets ((1)–(4)) show the fluorescence decays at different time points indicated by the arrows (the intensity scale is linear). (b) Fitting curves to the normalized decays. | ||
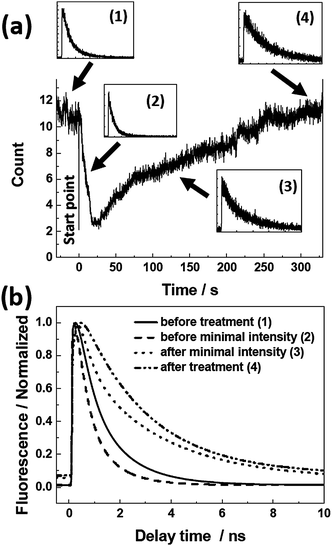
Fluorescence properties of the aggregates on treated glass were also studied during the vapor treatment (Fig. 5). The fluorescence intensity initially decreased, but to a much smaller extent than that observed on the untreated glass (Fig. 4). After a while the intensity started to increase until saturated at a level ∼10 times higher than the lowest intensity. If we compare the vapor treatment experiments on the untreated (Fig. 3) and treated (Fig. 5) glasses, there are clearly some similarities. The fluorescence intensity trace on the treated glass is very similar to the last part of that on the untreated glass (from 80 s to 500 s in Fig. 3). The spectra and fluorescence decays also show similar trends at the corresponding time intervals. This means that the treatment of the substrate with water imposes the same effect on the aggregate fluorescence as the initial exposure to water vapor of the sample prepared on the untreated glass. Obviously, the water-rinsing process saturates the glass surface with water molecules that are not removed by the subsequent drying procedure. Such small amount of residual water molecules can change optical properties, showing the sensitivity of PBI aggregation to traces of water.
 | ||
| Fig. 5 (a) Fluorescence intensity trace of an individual J-aggregate chain on treated glass. Insets ((1)–(4)) show the fluorescence decays at different time points indicated by the arrows (the intensity scale is linear). (b) Fitting curves to the normalized decays. | ||
Discussion
Considering the water rinsing process, the general surface properties of the such treated glass are not expected to be changed very much in comparison with the untreated glass. Therefore the observed difference of the optical properties of the attached dye aggregates is suggested to be due to the residual water rather than the hydrophobicity of the surface. To prove it, the water contact angles were measured for both surfaces. The values of 45° and 43° were obtained for the treated and untreated substrates, respectively (data not shown), showing that the hydrophobicity remains the same after water rinsing. The conclusion that the effects come from water and not from the surface is also supported by the vapor treatment experiments in which the water vapor can also change the properties of the PBI J-aggregates in a similar manner without changing the surface.The changes of the optical properties of PBI 1 J-aggregates observed during the water vapor treatment indicate that resonance coupling between monomers is suppressed. At a first glance it may seem as if the aggregate is dissociated into monomers. However, no isoemissive point was observed in our case, which means that this process is more complicated than a single-step dissociation from the J-aggregate to monomers. Note that here by dissociation we mean the loss of interaction between monomers, which is concluded from the optical properties. In order to alter dramatically the intermolecular interactions it is enough to change the positions of the monomers by a fraction of a nanometre. Obviously such small re-arrangements cannot be observed by fluorescence microscopy. Therefore, the same thread-like shape of the aggregates was seen by fluorescence microscopy (Fig. 2a and d) and only a small difference was observed in the AFM images within an ∼10 nm scale (Fig. 2c and f). Due to the intensity decrease and the absence of any spectral changes during the first period of water vapor treatment, we propose that an intermediate state is formed which exhibits a very low fluorescence quantum yield. Aggregates in this intermediate state can further dissociate into monomers when more water molecules are available. Both the intensity traces and the lifetime results suggest that the intermediate state corresponds to excitonically coupled PBI 1 monomers possessing decreased fluorescence yield due to the formation of quenching sites. The water molecules themselves cannot quench fluorescence, but they can induce molecular reorganization in J-aggregates.26,27 Humidity has already been reported to have a strong effect on the morphology of Langmuir–Blodgett-deposited dye monolayers,28–30 arrangement of the dye molecules within aggregates,26,31,32 and even the type of aggregation.27 For the PBI 1 J-aggregates, water molecules will coordinate at the imide hydrogen-bonding sites and concomitantly disrupt the hydrogen-bonded chain of dye molecules. Initially this coordination of water molecules is supposed to occur at defect sites such as missing of monomers in a double string aggregate, as illustrated in Fig. 6. The remaining π–π-contacts between PBI 1 molecules are, however, known to favor H-type excitonic coupling.11 H-type coupling plays an important role in electronic properties of majority of densely packed light-absorbing materials like e.g.conjugated polymers and molecular crystals.33 According to the exciton theory, H-aggregates are usually completely non-emissive and can act as quenchers. Each quencher (H-aggregate in this case) can efficiently quench an extended aggregate chain (up to 70 nm) due to the efficient exciton migration in the J-aggregated intact chain.23,34 The existence of easily accessible defects in PBI 1 J-aggregates is plausible since they possess quite a high level of disorder manifesting in broad spectral lines and short exciton coherent lengths (3–4 monomers at room temperature).35 The formation of H-aggregated sites can also occur at the end of the aggregate chain where the chain has a similar structure to the defect discussed above.
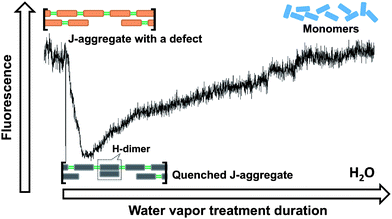 | ||
| Fig. 6 Possible reorganization of PBI 1 molecules in a J-aggregate during the water vapor treatment. | ||
Analysis of the fluorescence kinetic data supports this interpretation. The kinetic data reveal that during the first period of the vapor treatment (0–25 s, Fig. 4) the integrated fluorescence intensity drops as a result of the decrease of both the initial amplitude and the fluorescence lifetime. Likewise, the increase of the fluorescence intensity during the following time interval (25–330 s, Fig. 4) is also a combination of an increasing lifetime (up to ∼3 ns at the end of the treatment) and an increase of the initial amplitude of the signal. The lifetime of 3 ns as well as the spectral shape and maximum of the fluorescence spectrum (Fig. 2e) are unambiguous signatures of PBI 1 monomers. Accordingly, the change of the fluorescence intensity within the time interval between 25 and 330 s can be attributed to the water vapor induced disintegration of the hydrogen-bonded PBI chain, leading to PBI monomers without significant resonance interaction and lack of exciton transport capabilities.
Overall, this study indicates that a small amount of water molecules can induce great changes of the exciton coupling in the PBI 1 J-aggregate. Considering the spectrum in Fig. 2e and the intensity trace in Fig. 5, we can conclude that the PBI 1 aggregates deposited at the treated surface adopt different internal organization than the same PBI 1 aggregates deposited on the untreated surface. While the spectrum shown in Fig. 2e indicates that the observed fluorescence was indeed due to dissociated monomers, we assume that the most part of the dyes, however, was still arranged in the form of intact J-aggregates. However their (J-aggregates) fluorescence was quenched by a rapid exciton migration to trapping sites (presumably H-dimers). These intact J-aggregates further dissociated into monomers when the water vapor was introduced into the sample chamber. Thus the fluorescence intensity significantly increased as shown in Fig. 5, with the spectrum remaining the same. As mentioned above, for the chosen excitation wavelength the absorption cross-section of the PBI 1 monomer is ∼2 times higher than that of the J-aggregate. Accordingly, the intensity should be two times higher after the treatment if the aggregates were totally dissociated. However, similar fluorescence intensities were observed before and after the treatment as shown in Fig. 3 and 4. This can be due to the bleaching effect. The J-aggregates are quite stable but the monomers can be bleached easier after long laser light exposure.
Conclusion
In summary, we were able to observe by fluorescence microscopy at the individual aggregate level how water molecules changed the balance between π–π stacking and hydrogen-bonding interactions of PBI molecules, leading to structural re-arrangements in the aggregates. Initially a transition from highly fluorescent J-aggregates possessing clear excitonic properties to dark, non-fluorescent disordered aggregates was observed, which can be attributed to the formation of quenching sites upon interaction of water molecules with defect sites. The subsequent slower kinetic process leading to highly emissive ensembles of non-interacting dye molecules relates to the disintegration of the hydrogen-bonded supramolecular chain. These results reveal that an intact self-assembled dye aggregate is quite robust,36 and accordingly can be disassembled in a controlled way with concomitant changes of its excitonic properties. Such features are obviously important for applications of fluorescent dye aggregates. In addition, monitoring the fluorescent response of a single object to vapors might be an interesting approach for the elucidation of biological nanosystems or construction of sensory devices.Acknowledgements
This study was financially supported by The Swedish Research Council, The Royal Physiographic Society in Lund, the Knut & Alice Wallenberg Foundation, the Crafoord Foundation, and the Carl Trygger Foundation. Y.T. thanks the Swedish Institute for a postdoctoral scholarship. I.G.S. is grateful for a Linnaeus Grant to Lund Laser Centre. This work has been supported by the European Science Foundation within COST Action D35. F.W. thanks the Deutsche Forschungsgemeinschaft (DFG) for financial support within research training school 1221 “Control of electronic properties in aggregated π-conjugated molecules”.Notes and references
- E. Jelley, Nature, 1936, 138, 1009 CrossRef CAS.
- G. Scheibe, Angew. Chem., 1936, 49, 563 CAS.
- F. Würthner, T. E. Kaiser and C. R. Saha-Möller, Angew. Chem., Int. Ed., 2011, 50, 3376 CrossRef.
- S. Kirstein and S. Dähne, Int. J. Photoenergy, 2006, 1 Search PubMed , Article ID 20363.
- D. Möbius, Adv. Mater., 1995, 7, 437 CrossRef.
- R. J. Cogdell and J. Köhler, Biochem. J., 2009, 422, 193 CrossRef CAS.
- T. S. Balaban, H. Tamiaki and A. R. Holzwarth, Top. Curr. Chem., 2005, 258, 1 CrossRef CAS.
- A. Pawlik, S. Kirstein, U. DeRossi and S. Dähne, J. Phys. Chem. B, 1997, 101, 5646 CrossRef CAS.
- C. Spitz and S. Dähne, Int. J. Photoenergy, 2006, 2006, 1 CrossRef , Article ID 84950.
- I. G. Scheblykin, O. Y. Sliusarenko, L. S. Lepnev, A. G. Vitukhnovsky and M. Van der Auweraer, J. Phys. Chem. B, 2001, 105, 4636 CrossRef CAS.
- F. Würthner, Chem. Commun., 2004, 1564 RSC.
- X. Zhan, A. Facchetti, S. Barlow, T. J. Marks, M. A. Ratner, M. R. Wasielewski and S. R. Marder, Adv. Mater., 2011, 23, 268 CrossRef CAS.
- F. Würthner and M. Stolte, Chem. Commun., 2011, 47, 5109 RSC.
- K. Y. Law, Chem. Rev., 1993, 93, 449 CrossRef CAS.
- C. W. Tang, Appl. Phys. Lett., 1986, 48, 183 CrossRef CAS.
- L. Schmidt-Mende, A. Fechtenkotter, K. Müllen, E. Moons, R. H. Friend and J. D. MacKenzie, Science, 2001, 293, 1119 CrossRef CAS.
- S. Ghosh, X.-Q. Li, V. Stepanenko and F. Würthner, Chem.–Eur. J., 2008, 14, 11343 CrossRef CAS.
- T. E. Kaiser, V. Stepanenko and F. Würthner, J. Am. Chem. Soc., 2009, 131, 6719 CrossRef CAS.
- T. E. Kaiser, H. Wang, V. Stepanenko and F. Würthner, Angew. Chem., Int. Ed., 2007, 46, 5541 CrossRef CAS.
- W. E. Moerner and L. Kador, Phys. Rev. Lett., 1989, 62, 2535 CrossRef CAS.
- M. Orrit and J. Bernard, Phys. Rev. Lett., 1990, 65, 2716 CrossRef CAS.
- E. Lang, A. Sorokin, M. Drechsler, Y. V. Malyukin and J. Köhler, Nano Lett., 2005, 5, 2635 CrossRef CAS.
- H. Z. Lin, R. Camacho, Y. X. Tian, T. E. Kaiser, F. Würthner and I. G. Scheblykin, Nano Lett., 2010, 10, 620 CrossRef CAS.
- H. Z. Lin, S. R. Tabaei, D. Thomsson, O. Mirzov, P. O. Larsson and I. G. Scheblykin, J. Am. Chem. Soc., 2008, 130, 7042 CrossRef CAS.
- When adsorbed on a glass surface the lifetime of both PBI monomers and aggregates becomes shorter than reported for the corresponding solutions in ref. 19. This can be due to fluorescence quenching by the glass surface.
- R. Shinozaki and T. Nakato, Langmuir, 2004, 20, 7583 CrossRef CAS.
- K. Okuyama, M. Ikeda, S. Yokoyama, Y. Ochiai, Y. Hamada and M. Shimomura, Chem. Lett., 1988, 1013 CrossRef CAS.
- Y. L. E. Chen and J. N. Israelachvili, J. Phys. Chem., 1992, 96, 7752 CrossRef CAS.
- Y. L. E. Chen, M. L. Gee, C. A. Helm, J. N. Israelachvili and P. M. Mcguiggan, J. Phys. Chem., 1989, 93, 7057 CrossRef CAS.
- H. Yao, Z. M. Ou, Y. Morita and K. Kimura, Colloids Surf., A, 2004, 236, 31 CrossRef CAS.
- T. Seki, T. Fukuchi and K. Ichimura, Langmuir, 2000, 16, 3564 CrossRef CAS.
- F. Nüesch, J. E. Moser, V. Shklover and M. Grätzel, J. Am. Chem. Soc., 1996, 118, 5420 CrossRef.
- F. C. Spano, Acc. Chem. Res., 2010, 43, 429 CrossRef CAS.
- H. Marciniak, X.-Q. Li, F. Würthner and S. Lochbrunner, J. Phys. Chem. A, 2011, 115, 648 CrossRef CAS.
- T. E. Kaiser, I. G. Scheblykin, D. Thomsson and F. Würthner, J. Phys. Chem. B, 2009, 113, 15836 CrossRef CAS.
- E. Krieg, H. Weissman, E. Shirman, E. Shimoni and B. Rybtchinski, Nat. Nanotechnol., 2011, 6, 141 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Fluorescence spectra of PBI 1 monomers on treated and untreated glasses. See DOI: 10.1039/c1nr10973a |
| This journal is © The Royal Society of Chemistry 2012 |