Zinc– and palladium–porphyrin based turnstiles†‡
Thomas
Lang
,
Ernest
Graf
*,
Nathalie
Kyritsakas
and
Mir Wais
Hosseini
*
UMR UDS-CNRS 7140, Université de Strasbourg, Institut Le Bel, 4 rue Blaise Pascal, CS90032 67081 Strasbourg, France. E-mail: hosseini@unistra.fr; egraf@unistra
First published on 25th September 2012
Abstract
The design and synthesis of two new Zn(II) and Pd(II) strapped-porphyrin based molecular turnstiles have been achieved. These molecules are based on a porphyrin backbone as a stator and a handle as a rotor. The junction between the two parts is ensured by covalent bonds using two opposite meso positions. Both compounds have been characterised in solution by NMR spectroscopy and in the solid state by X-ray diffraction on a single crystal. The dynamic of the two systems have been investigated in solution by 1- and 2-D NMR techniques such as ROESY. The switching between the open and closed states is described. Whereas for the Pd(II) complex, the handle freely rotates around the stator, for the Zn(II) analogue owing to the presence of a water molecule bound to the zinc atom and hydrogen bonded to the pyridyl moiety of the rotor, the rotation is blocked. The unlocking of the rotational movement was achieved using an external agent, mainly dimethylaminopyridine as a strong coordinating ligand.
Introduction
The design and synthesis of molecular architectures for which intramolecular movements can be controlled by external stimuli are a topic of interest (for review articles see ref. 1–8). Many examples of rotaxanes9 and catenanes10 based systems have been reported. Molecular motors functioning with thermal11 or photochemical12 processes have been published. Molecular cars13 and wheelbarrows14 have also been described. Among the above-mentioned systems, molecular turnstiles15 form a class of dynamic architectures subject to rotational processes between a fixed (stator) and a mobile (rotor) part. For this type of molecules, the switching between an open and a closed state by an external stimulus is challenging and of interest. We have previously reported examples of turnstiles based either on Sn–porphyrin derivatives16,17 or on a strapped-porphyrin for which the rotor comprises a tridentate unit.18Here, we report on the design, synthesis, characterization, both in solution and in the crystalline phase, and on the switching between the open and closed states of molecular turnstiles based on strapped-porphyrins19 composed of a free base meso tetraaryl porphyrin and its zinc20 or palladium complexes and a covalently attached handle bearing a pyridyl moiety as a monodentate coordinating site.
Results and discussion
The turnstile reported here is the strapped porphyrin 1 (Scheme 1). Its design is based on a meso tetraaryl porphyrin as a stator and a strap as a rotor. It should be noted that the designation of the two parts as a stator and as a rotor is arbitrary and the rotational process is a relative movement of one of the two parts around the other. The rational behind the choice of the porphyrin backbone is based on its propensity to bind a large variety of metal centres by its tetraaza macrocyclic core and on available synthetic possibilities to incorporate the strap. Examples of strapped porphyrins bearing a phenanthroline unit as a handle have been reported.21 The rotor is a monodentate pyridyl coordinating unit bearing, in a symmetrical fashion, two tetraethyleneglycol moieties at positions 2 and 6. The pyridyl unit and the spacer are interconnected using ether junctions. The connection between the porphyrin and the strap is ensured by two covalent ether groups using the two meso positions 5 and 15 on the porphyrin. For the attachment of the handle to the aryl groups of the porphyrin, one may use the ortho, meta or para position. In order to avoid (i) a rotational barrier induced by the use of the ortho position, (ii) longer spacer units between the porphyrin backbone and the pyridyl moiety resulting from the improper angle imposed by the use of the para position, the junction is achieved in a symmetrical fashion using the meta position on both aryl groups in positions 5 and 15. The remaining two meso positions 10 and 20 are occupied by two benzonitrile groups connected to the porphyrin backbone using the position 4 of the aryl moiety. The nitrile groups at the meso positions were introduced as additional coordinating sites in order to explore the possibility of locking the system by simultaneous binding of external metal cations by both the peripheral site and the pyridyl moiety of the handle. Their presence does not interfere with the present study. | ||
| Scheme 1 Strapped porphyrin 1, its Zn(II) and Pd(II) complexes 2 and 3 respectively, and the water complex 2·H2O and the assignment of H atoms. | ||
The operational design of the turnstile (Fig. 1) is based on: (i) free rotation (open state, Fig. 1a) of the rotor around the stator as a consequence of the absence of specific interactions between the two parts, (ii) introduction of the Zn(II) cation within the macrocyclic coordinating moiety of the strapped porphyrin 1 (Fig. 1b) affording the neutral complex 2 (Scheme 1). Owing to the propensity of Zn2+ cation to adopt mainly a square based pyramidal geometry when complexed by the tetraaza core of the porphyrin, the coordination of the pyridyl unit of the rotor should lock the rotation process leading thus to the closed state of the turnstile (Fig. 1b). In order to prove the latter statement, the Zn2+ cation may be replaced by Pd(II) (complex 3, Scheme 1) adopting the square planar geometry without extension of its coordination sphere. In that case, the locking process cannot take place and thus the rotation of the rotor around the stator should not be altered (Fig. 1c).
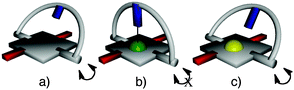 | ||
| Fig. 1 Schematic representation of strapped-porphyrins: (a) free base (open state), (b) Zn–porphyrin (closed state), (c) Pd–porphyrin (open state). | ||
The synthetic strategy adopted for the preparation of the strapped porphyrin 1 (Scheme 2) was based on the condensation of the dipyrrylmethane 1322 and the α, ω dialdehyde 11. The latter was prepared from the commercially available 3-hydroxybenzaldehyde 14, tetraethyleneglycol 4 and 2,6-pyridine diol 6.
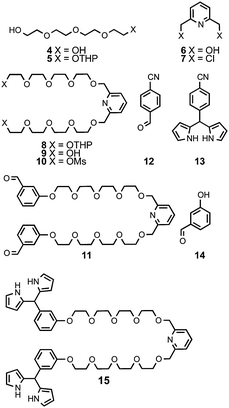 | ||
| Scheme 2 | ||
Compound 4 was first converted at room temperature in the presence of a catalytic amount of pyridinium para-toluenesulfonate (PPTS) into its mono-DHP protected derivative 523 upon treatment with dihydropyrane (DHP) in CHCl3 (34% yield). The latter was then condensed in the presence of NaH in refluxing THF with the dichloro compound 724 affording the compound 825 in 54% yield. Compound 7 was prepared in 82% yield in THF at room temperature upon treatment of 6 with SOCl2. The bis-OTHP derivative 8 was then deprotected under acidic conditions affording the diol 9 in 88% yield. The latter was quantitatively activated into its dimesylate derivative 10. The handle 11 was obtained in the presence of potassium carbonate in 96% yield in refluxing CH3CN upon reaction with 3-hydroxybenzaldehyde 14.
Dipyrrylmethane 1322 was obtained in 63% yield upon reaction of the aldehyde 12 with pyrrole in the presence of TFA. Condensation, in a CH2Cl2–EtOH mixture and in the presence of TFA, of the dialdehyde 11 with dipyrrylmethane 13 followed by oxidation in THF and in the presence of Et3N using DDQ,26 afforded strapped-porphyrin 1 in 2% yield.
In order to increase the yield of the final step, another synthetic strategy was explored. The compound 11 was converted in 66% into its bisdipyrrylmethane derivative 15 by reaction with an excess of pyrrole in the presence of TFA. Unfortunately, this strategy did not enhance the yield of the porphyrin formation. Indeed, the condensation of 15 in the presence of TFA with the aldehyde 12 in a CH2Cl2–EtOH mixture followed by oxidation using DDQ26 in the presence of Et3N in THF afforded the porphyrin derivative 1 in 1% yield.
The Zn–porphyrin complex 2 was quantitatively obtained in a CH2Cl2–MeOH mixture and at room temperature, upon treatment of 1 with Zn(OAc)2. The Pd–porphyrin complex 3 was obtained in 66% yield in a refluxing CHCl3–MeOH mixture, upon addition of Pd(OAc)2 to a solution of the porphyrin 1.
The assignment of all hydrogen atoms of the strapped porphyrins 1–3 was achieved at 25 °C in CD2Cl2 by 1- and 2-D NMR COSY and ROESY experiments. Using the same technique, the dynamics of compounds 1–3 in solution (CD2Cl2) was also studied at 25 °C.
For both porphyrins 1 and 3, a splitting of hydrogen atoms of the benzonitrile moieties Ha, and Hb into Ha, 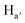 , and Hb, Hb (Scheme 1) was observed. This differentiation, resulting from the slow rotation of the handle around the porphyrin core within the NMR time scale, is in agreement with a previous study performed on an analogue system.18 The binding of the Pd(II) cation by the tetraaza core of the porphyrin has almost no influence on the H atoms of the handle. Indeed, when compared to the parent compound 1, signals corresponding to Hr and Hs of the pyridyl unit are only slightly shifted by 0.03 ppm and 0.06 ppm respectively. The same observation holds for Hu and Hv atoms belonging to the tetraethyleneglycol units which are shifted by 0.08 ppm and 0.04 ppm, respectively (Fig. 2a and c). These observations indicate the free rotation of the handle around the porphyrin stator defining thus the open state of the turnstile.
, and Hb, Hb (Scheme 1) was observed. This differentiation, resulting from the slow rotation of the handle around the porphyrin core within the NMR time scale, is in agreement with a previous study performed on an analogue system.18 The binding of the Pd(II) cation by the tetraaza core of the porphyrin has almost no influence on the H atoms of the handle. Indeed, when compared to the parent compound 1, signals corresponding to Hr and Hs of the pyridyl unit are only slightly shifted by 0.03 ppm and 0.06 ppm respectively. The same observation holds for Hu and Hv atoms belonging to the tetraethyleneglycol units which are shifted by 0.08 ppm and 0.04 ppm, respectively (Fig. 2a and c). These observations indicate the free rotation of the handle around the porphyrin stator defining thus the open state of the turnstile.
 | ||
| Fig. 2 A portion (2.1–9.1 ppm) of the 1H-NMR spectra (CD2Cl2, 400 MHz, 25 °C) of 1 (a), 2 (b) and 3 (c). * and ¤ correspond to residual CHCl3 and MeOH, respectively. For assignment of H atoms see Scheme 1. | ||
This statement was further confirmed by a 2-D ROESY experiment at room temperature which revealed only the expected correlations between Hk and Hj and Hg due to spatial proximities resulting from the atomic connectivity in the free strapped porphyrin 1 and its Pd complex 3 (Fig. 3a and b).
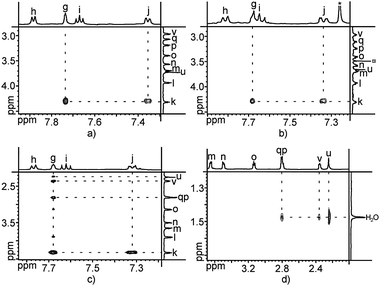 | ||
| Fig. 3 Portions of the 2-D ROESY correlations for 1 (a), 3 (b) and 2 (c and d). * and ¤ correspond to residual chloroform and methanol, respectively. For attribution of H atoms see Scheme 1. | ||
For the Zn–porphyrin turnstile 2, the C2v symmetry of the molecule is conserved. The 1H-NMR investigations revealed, as expected, a drastic upfield shift of Hu and Hv atoms by 1.32 ppm and 0.53 ppm, respectively, implying the locking of the handle and the positioning of this portion of the handle within the anisotropic cone of the porphyrin backbone. The observed splitting increase between Hb and  signals is another argument in favour of a blocked rotation. However, the pyridine Hr and Hs hydrogen atoms were found to be downfield shifted by 0.35 ppm and 0.85 ppm, respectively (Fig. 2b). This observation is not in agreement with a direct binding of the Zn(II) cation located within the centre of the porphyrin stator by the N atom of the pyridyl unit of the handle. Indeed, if this was the case, one would expect upfield shifts of Hr and Hs signals. Thus, the closed state of the turnstile must result from an indirect interaction between the handle and the stator. The 2-D ROESY sequence at room temperature revealed, in addition to the expected correlations between Hk and Hj and Hg, as also observed for 1 (Fig. 3a) and 3 (Fig. 3b), new correlations between Hg of the meso aromatic groups and Hu, Hv and Hq belonging to the handle (Fig. 3c). Furthermore, it also revealed the presence of correlations between Hu, Hv and Hq and a water molecule (Fig. 3d). This observation is consistent with the coordination of a water molecule by the Zn cation and interacting with the handle through a H-bond with the pyridyl moiety affording the complex 2·H2O (Scheme 1). This proposal also explains why the proton signals of Hr and Hs of the pyridyl unit are slightly downfield shifted. This hypothesis is substantiated in the crystalline phase by the structural investigation on a single crystal presented hereafter.
signals is another argument in favour of a blocked rotation. However, the pyridine Hr and Hs hydrogen atoms were found to be downfield shifted by 0.35 ppm and 0.85 ppm, respectively (Fig. 2b). This observation is not in agreement with a direct binding of the Zn(II) cation located within the centre of the porphyrin stator by the N atom of the pyridyl unit of the handle. Indeed, if this was the case, one would expect upfield shifts of Hr and Hs signals. Thus, the closed state of the turnstile must result from an indirect interaction between the handle and the stator. The 2-D ROESY sequence at room temperature revealed, in addition to the expected correlations between Hk and Hj and Hg, as also observed for 1 (Fig. 3a) and 3 (Fig. 3b), new correlations between Hg of the meso aromatic groups and Hu, Hv and Hq belonging to the handle (Fig. 3c). Furthermore, it also revealed the presence of correlations between Hu, Hv and Hq and a water molecule (Fig. 3d). This observation is consistent with the coordination of a water molecule by the Zn cation and interacting with the handle through a H-bond with the pyridyl moiety affording the complex 2·H2O (Scheme 1). This proposal also explains why the proton signals of Hr and Hs of the pyridyl unit are slightly downfield shifted. This hypothesis is substantiated in the crystalline phase by the structural investigation on a single crystal presented hereafter.
The solid state structures of both compound 2 and 3 were studied by X-ray diffraction on single crystals obtained upon slow diffusion of pentane into a CH2Cl2 solution of 2 (Fig. 4) or 3 (Fig. 5).
 | ||
| Fig. 4 Solid state structure of compound 2·H2O. One of the two tetraethyleneglycol units connecting the handle to the porphyrin is found to be disordered. Hydrogen atoms and solvent molecules are omitted for clarity. For bond distances and angles, see text. | ||
 | ||
| Fig. 5 Solid state structure of compound 3. Hydrogen atoms and solvent molecules are omitted for clarity. One of the ethyleneglycol units is found to be disordered. For bond distances and angles, see text. | ||
For the Zn–porphyrin 2·H2O complex, the structural study revealed that the crystal (triclinic, P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ) in addition to the complex 2 contains water and dichloromethane molecules (Fig. 4). No noticeable deformation of the porphyrin backbone is observed. The zinc cation is located above the porphyrin mean plane (0.441 Å). The coordination geometry around the metallic centre is a slightly distorted square based pyramid. Its coordination sphere is composed of all four N atoms of the porphyrin core with Zn–N distances in the 2.063(3)–2.083(3) Å range and N–Zn–N angles of 160.02(12)–160.97(12)° (N–Zn–Ntrans), 87.67(11)–89.05(11)° (N–Zn–Ncis). The water molecule occupies the apical position with the Zn–O distance of 2.120(3) Å and N–Zn–O angles of 98.53(11)–100.49(11)°. The four meso substituents are not coplanar but tilted with respect to the porphyrin mean plane with CCCC dihedral angles of −67.5° and −71.2° for the aryl units of the bridging handle and of −80.2° and 59.7° for the benzonitrile groups. One of the two tetraethyleneglycol units connecting the handle to the porphyrin is found to be disordered. Interestingly, the water molecule is hydrogen bonded to the pyridine nitrogen atoms of the handle with a O–N bond distance of 2.915(4) Å and NHO angles of 165(4)°.
) in addition to the complex 2 contains water and dichloromethane molecules (Fig. 4). No noticeable deformation of the porphyrin backbone is observed. The zinc cation is located above the porphyrin mean plane (0.441 Å). The coordination geometry around the metallic centre is a slightly distorted square based pyramid. Its coordination sphere is composed of all four N atoms of the porphyrin core with Zn–N distances in the 2.063(3)–2.083(3) Å range and N–Zn–N angles of 160.02(12)–160.97(12)° (N–Zn–Ntrans), 87.67(11)–89.05(11)° (N–Zn–Ncis). The water molecule occupies the apical position with the Zn–O distance of 2.120(3) Å and N–Zn–O angles of 98.53(11)–100.49(11)°. The four meso substituents are not coplanar but tilted with respect to the porphyrin mean plane with CCCC dihedral angles of −67.5° and −71.2° for the aryl units of the bridging handle and of −80.2° and 59.7° for the benzonitrile groups. One of the two tetraethyleneglycol units connecting the handle to the porphyrin is found to be disordered. Interestingly, the water molecule is hydrogen bonded to the pyridine nitrogen atoms of the handle with a O–N bond distance of 2.915(4) Å and NHO angles of 165(4)°.
For the Pd–porphyrin complex 3, the crystal (triclinic, P) is composed of the porphyrin 3, pentane and H2O solvent molecules (Fig. 5). The latter occupy in the crystal the empty space between compounds 3 without any specific interactions. The porphyrin backbone is not distorted. The palladium cation, located almost in the centre of the porphyrin core, is tetracoordinated. The coordination geometry around the metallic centre is almost square planar with Pd–N distances in the 1.995(9)–2.024(10) Å range and N–Pd–N angles of 178.3(4)–178.6(4)° (N–Pd–Ntrans), 89.7(4)–90.6(4)° (N–Pd–Ncis). The meso substituents are tilted with respect to the porphyrin mean plane with CCCC dihedral angle of −65.6° and −57.6° for the aryl units of the bridging handle and −75.0° and 61.0° for the benzonitrile groups. One of the glycol units is found to be disordered over two positions.
In order to unlock the turnstile, 1–10 equivalents of para-dimethylaminopyridine (DMAP), a competitive ligand, was added to 2 and the process monitored by 1H-NMR in CD2Cl2 at 25 °C (Fig. 6). As expected, upon addition of one equivalent of DMAP (Fig. 6b), the open state of the turnstile was restored. Indeed, by considering the “free-base” strapped-porphyrin 1 as a reference (Fig. 6d), the presence of DMAP caused a shift of all proton signals towards those observed for 1. In particular, Hr and Hs hydrogen atoms of the pyridyl moiety were upfield shifted by 0.16 ppm and 0.65 ppm respectively, whereas Hu and Hv signals were downfield shifted by 1.30 ppm and 0.55 ppm respectively. Furthermore, Hb and  signals were found to be less differentiated. Upon further addition of DMAP, no additional shift of signals of the turnstile was observed (see Fig. 6c for addition of 2 eq. of DMAP). Dealing with DMAP, due to the fast exchange dynamics, its signals appeared to be broadened. However, for the 1/1 mixture of DMAP/2, in agreement with the coordination of DMAP to the Zn cation, the signal corresponding to the CH3 groups was found to be upfield shifted by 0.53 ppm. Upon increasing the DMAP/2 ratio from 1 to 10, all signals of DMAP became narrower and downfield shifted.
signals were found to be less differentiated. Upon further addition of DMAP, no additional shift of signals of the turnstile was observed (see Fig. 6c for addition of 2 eq. of DMAP). Dealing with DMAP, due to the fast exchange dynamics, its signals appeared to be broadened. However, for the 1/1 mixture of DMAP/2, in agreement with the coordination of DMAP to the Zn cation, the signal corresponding to the CH3 groups was found to be upfield shifted by 0.53 ppm. Upon increasing the DMAP/2 ratio from 1 to 10, all signals of DMAP became narrower and downfield shifted.
 | ||
| Fig. 6 Portion of the 1H NMR spectra (CD2Cl2, 300 MHz, 25 °C) of 2·H2O (a) in the presence of 0 eq. (a), 1 eq. (b) and 2 eq. (c) of DMAP and of the parent compound 1 (d). * corresponds to signals of coordinated DMAP. For assignment of H atoms see Scheme 1. | ||
Conclusion
In conclusion, a new strapped-porphyrin based molecular turnstile was designed, synthesized and characterised both in solution and in the solid state. The turnstile is composed of a porphyrin backbone (stator) equipped with a handle bearing a pyridyl unit as a monodentate coordinating site (rotor). In the absence of Zn(II) cation within the tetraaza core of the porphyrin, the rotor freely rotates around the stator (open state). In the presence of Pd(II) within the porphyrin core and adopting a square planar geometry, again the free rotation of the rotor around the stator is observed. In the presence of Zn(II), adopting a square based pyramidal geometry, in contrast with the initial design based on direct coordination of Zn cation by the pyridyl unit, the rotation was found to be locked (closed state) owing to the formation of a H-bond between a water molecule coordinated to the metal centre and the pyridyl moiety of the handle. The addition of DMAP, a competitive ligand, to the turnstile in its closed state leads to the open state of the turnstile. The use of rotors bearing tridentate binding site and other metal centres either bound to the porphyrin and/or to the rotor is currently under investigation. Furthermore, taking advantage of the presence of the peripheral nitrile coordinating sites, the locking of the turnstile using additional metal centres is also currently explored.Experimental
General procedure
THF and triethylamine were distilled over sodium and KOH, respectively. Analytical grade of CH2Cl2, CHCl3, EtOH and MeOH was used without further purification. 1H- and 13C-NMR spectra were acquired at 25 °C on either a Bruker AV 300, Bruker AV 400 or a Brucker AV 500 spectrometers. Deuterated solvents were used as the lock and residual non deuterated solvents as the internal references. Mass spectrometry analyses were performed by the Service de Spectrometrie de Masse, University of Strasbourg.X-ray crystal-structure analysis
Data were collected on a Bruker APEX8 CCD diffractometer equipped with an Oxford Cryosystem liquid N2 device at 173(2) K using a molybdenum microfocus sealed tube generator with mirror-monochromated Mo-Kα radiation (λ = 0.71073 Å), operated at 50 kV/600 mA. The structure was solved using SHELXS-97 and refined by fullmatrix least-squares on F2 using SHELXL-97 with anisotropic thermal parameters for all non-hydrogen atoms.27 The hydrogen atoms were introduced at calculated positions and not refined (riding model).Crystals of 2 and 3 were obtained in a crystallization tube (0.5 × 20 cm) at room temperature upon slow diffusion of pentane into a solution of the complex in CH2Cl2 through a buffer layer of pentane/CH2Cl2 (1/1).
Crystallographic data for
2: C138.5H131ClN14O23Zn2, M = 2525.81, triclinic, P, a = 13.5277(4) Å, b = 15.3111(7) Å, c = 17.7296(5) Å, α = 86.083(3)°, β = 67.796(2)°, γ = 77.863(3)°, U = 3323.7(2) Å3, Z = 1, μ = 0.454 mm−1, refls measured: 34![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 801, independent refls: 14673 [Rint = 0.0240], final R indices [I > 2σ (I)]: R1 = 0.0782, wR2 = 0.2357, R indices (all data): R1 = 0.0969, wR2 = 0.2528, GOF on F2: 1.063.
801, independent refls: 14673 [Rint = 0.0240], final R indices [I > 2σ (I)]: R1 = 0.0782, wR2 = 0.2357, R indices (all data): R1 = 0.0969, wR2 = 0.2528, GOF on F2: 1.063.
Crystallographic data for
3: C148H156N14O23Pd2, M = 2711.67, triclinic, P, a = 14.8196(12) Å, b = 17.1292(15) Å, c = 17.1532(17) Å, α = 63.953(3)°, β = 84.648(3)°, γ = 64.883(3)°, U = 3518.9(5) Å3, Z = 1, μ = 0.328 mm−1, refls measured: 12750, independent refls: 11654 [Rint = 0.0378], final R indices [I > 2σ (I)]: R1 = 0.1132, wR2 = 0.2997, R indices (all data): R1 = 0.1510, wR2 = 0.3255, GOF on F2: 1.416. Owing to the weak diffraction power of the crystal, the data collection was performed with a theta value of 25°.
Synthesis
Compounds 724 and 1322 have been prepared following reported procedures.1H-NMR (CD2Cl2, 400 MHz) δ (ppm): −2.94 (s, 2H, NHy), 2.88 (m, 4H, OCH2v), 3.00 (m, 4H, OCH2q), 3.14 (m, 4H, OCH2p), 3.34 (m, 4H, OCH2o), 3.51 (m, 4H, OCH2n), 3.57 (s, 4H, OCH2u), 3.64 (m, 4H, OCH2m), 3.89 (m, 4H, OCH2l), 4.31 (m, 4H, OCH2k), 6.15 (d, 2H, Pyr, 3J = 7.5 Hz), 6.46 (t, 1H, Pys, 3J = 7.5 Hz), 7.36 (dd, 2H, Arj, 3J = 8.5 Hz, 4J = 2.0 Hz), 7.69 (t, 2H, Ari, 3J = 8.0 Hz), 7.76 (m, 2H, Arg), 7.88 (d, 2H, Arh, 3J = 7.5 Hz), 8.08 (d, 4H, Ara, 3J = 8.5 Hz), 8.33 (m, 4H, Arb), 8.76 (d, 4H, β-pyrc, 3J = 4.5 Hz), 8.94 (d, 4H, β-pyrd, 3J = 4.5 Hz). 13C-NMR (CD2Cl2, 100 MHz) δ (ppm): 68.0, 69.7, 69.9, 70.0, 70.4, 70.5, 70.7, 71.0, 73.3, 112.0, 114.5, 119.1, 119.2, 120.7, 122.2, 127.7, 128.0, 130.7, 131.1, 132.3, 135.4, 136.3, 143.1, 147.0, 157.1, 157.7. MS (ESI) for C69H65N7O10: 1152.30 g mol−1. m/z (M + Na+): calculated = 1174.469; measured = 1174.497.
1H-NMR (CD2Cl2, 400 MHz) δ (ppm): 2.25 (s, 4H, OCH2u), 2.35 (m, 4H, OCH2v), 2.79 (m, 4H, OCH2p), 2.81 (m, 4H, OCH2q), 3.13 (m, 4H, OCH2o), 3.49 (m, 4H, OCH2n), 3.65 (m, 4H, OCH2m), 3.89 (m, 4H, OCH2l), 4.32 (m, 4H, OCH2k), 6.50 (d, 2H, Pyr, 3J = 7.5 Hz), 7.31 (m, 3H, Pys, Arj), 7.62 (t, 2H, Ari, 3J = 8.0 Hz), 7.68 (br, 2H, Arg), 7.77 (d, 2H, Arh, 3J = 7.5 Hz), 8.02 (m, 6H, Ara,b), 8.29 (d, 2H, Arb, 3J = 8.0 Hz), 8.77 (d, 4H, β-pyrc, 3J = 4.5 Hz), 8.95 (d, 4H, β-pyrd, 3J = 4.5 Hz). 13C-NMR (CD2Cl2, 100 MHz) δ (ppm): 68.6, 69.3, 69.6, 69.6, 70.0, 70.5, 70.6, 70.9, 71.0, 111.5, 114.4, 118.7, 119.5, 120.0, 121.1, 122.6, 127.6, 128.1, 130.5, 131.5, 132.8, 135.0, 135.4, 137.8, 144.4, 148.3, 149.6, 150.5, 156.3, 157.5.MS (ESI) for C69H63N7O10Zn: 1215.66 g mol−1. m/z (M + Na+): calculated = 1236.382; measured = 1236.400.
1H-NMR (CD2Cl2, 400 MHz) δ (ppm): 2.92 (m, 4H, OCH2v), 3.07 (m, 4H, OCH2), 3.20 (m, 4H, OCH2p), 3.37 (m, 4H, OCH2o), 3.53 (m, 4H, OCH2n), 3.65 (m, 8H, OCH2u,m), 3.89 (m, 4H, OCH2l), 4.30 (m, 4H, OCH2k), 6.18 (d, 2H, Pyr, 3J = 7.5 Hz), 6.52 (t, 1H, Pys, 3J = 7.5 Hz), 7.35 (dd, 2H, Arj, 3J = 8.0 Hz, 4J = 2.0 Hz), 7.68 (m, 4H, Arg,i), 7.82 (d, 2H, Arh, 3J = 7.5 Hz), 8.07 (m, 4H, Ara), 8.26 (m, 2H, Arb), 8.32 (m, 2H, Arb), 8.72 (d, 4H, β-pyrc, 3J = 5.0 Hz), 8.91 (d, 4H, β-pyrd, 3J = 5.0 Hz). 13C-NMR (CDCl3, 125 MHz) δ (ppm): 68.0, 69.7, 69.8, 70.0, 70.3, 70.5, 70.7, 70.9, 73.2, 111.5, 114.9, 118.5, 119.2, 119.2, 121.2, 121.5, 127.6, 128.0, 130.8, 130.9, 132.2, 134.7, 136.6, 140.8, 141.0, 142.2, 146.2, 156.5, 157.1. MS (ESI) for C69H63N7O10Pd: 1255.37 g mol−1. m/z (M + Na+): calculated = 1278.359; measured = 1278.358.
1H-NMR (CDCl3, 300 MHz) δ (ppm): 1.38–1.85 (m, 6H, CH2(THP)), 2.83 (br, 1H, OH), 3.40–3.85 (m, 18H, OCH2, OCH2(THP)), 4.58 (m, 1H, CH(THP)). 13C-NMR (CDCl3, 75 MHz) δ (ppm): 19.6, 25.6, 30.7, 61.7, 62.4, 66.8, 70.1, 70.7, 70.8, 73.1, 99.1. Anal.: calculated for C13H26O6: C = 56.10%; H = 9.42%; found: C = 56.53%; H = 9.23%.
1H-NMR (CDCl3, 300 MHz) δ (ppm): 1.47–1.87 (m, 12H, CH2 (THP)), 3.45–3.90 (m, 36H, OCH2k–v, OCH2 (THP)), 4.62 (m, 2H, CH (THP)), 4.68 (s, 4H, OCH2u), 7.39 (d, 2H, Pyr, 3J = 8.0 Hz), 7.72 (t, 1H, Pys, 3J = 8.0 Hz). 13C-NMR (CDCl3, 75 MHz) δ (ppm): 19.6, 25.6, 30.7, 62.4, 66.8, 70.4, 70.7, 70.8, 70.8, 74.0, 99.1, 120.1, 137.5, 157.9. Anal.: calculated for C33H57NO12: C = 60.07%; H = 8.71%; N = 2.12%; found: C = 59.95%; H = 9.07%; N = 1.88%.
1H-NMR (CDCl3, 300 MHz) δ (ppm): 2.83 (br, 2H, OH), 3.58–3.75 (m, 32H, OCH2k–v), 4.67 (s, 4H, OCH2u), 7.38 (d, 2H, Pyr, 3J = 8.0 Hz), 7.71 (t, 1H, Pys, 3J = 8.0 Hz). 13C-NMR (CDCl3, 75 MHz) δ (ppm): 61.8, 70.4, 70.5, 70.7, 70.8, 72.7, 74.0, 120.2, 137.6, 157.9. Anal.: calculated for C23H41NO10 0.5 C4H10O: C = 56.80%; H = 8.77%; N = 2.65%; found: C = 56.79%; H = 8.70%; N = 2.62%.
1H-NMR (CDCl3, 300 MHz) δ (ppm): 3.06 (s, 6H, CH3), 3.62–3.77 (m, 28H, OCH2v–l), 4.36 (m, 4H, OCH2k), 4.68 (s, 4H, OCH2u), 7.39 (d, 2H, Pyr, 3J = 8.0 Hz), 7.73 (t, 1H, Pys, 3J = 8.0 Hz). 13C-NMR (CDCl3, 75 MHz) δ (ppm): 37.9, 69.1, 69.4, 70.4, 70.7, 70.8, 73.9, 120.3, 137.8, 157.8. Anal.: calculated for C25H45NO14S2: C = 46.36%; H = 7.00%; N = 2.16%; found: C = 46.35%; H = 6.87%; N = 2.05%.
Compound 11
In a 500 mL two-necked round-bottom flask, 3-hydroxybenzaldehyde 14 (2.35 g, 19 mmol, 4 eq.) was dissolved in 200 mL of dry CH3CN. K2CO3 (2.65 g, 19 mmol, 4 eq.) was added and the reaction mixture stirred under argon at room temperature for 2 hours. A solution of compound 10 (3.11 g, 4.8 mmol, 1 eq.) in 100 mL of dry CH3CN was added via cannula. The reaction mixture was heated at reflux for 16 hours. The solvent was removed under vacuum and the oily residue was dissolved in CH2Cl2 (300 mL), washed with water (2 × 150 mL) and successively with solutions of NaOH (0.1 M, 150 mL), HCl (0.1 M, 150 mL) and brine (150 mL). The organic layer was dried over MgSO4 and the solvent removed under vacuum to yield the compound 11 (3.22 g, 96%) as a brown oil.1H-NMR (CDCl3, 300 MHz) δ (ppm): 3.65–3.75 (m, 24H, OCH2v–m), 3.87 (m, 4H, OCH2l), 4.18 (m, 4H, OCH2k), 4.67 (s, 4H, OCH2u), 7.19 (m, 2H, Ar), 7.35–7.44 (m, 8H, Ar, Pyr), 7.69 (t, 1H, Pys, 3J = 8.0 Hz), 9.95 (s, 2H, CHO). 13C-NMR (CDCl3, 75 MHz) δ (ppm): 67.9, 69.7, 70.4, 70.7, 70.8, 71.0, 74.0, 113.1, 120.1, 122.2, 123.7, 130.2, 137.9, 157.9, 159.5, 192.2.
1H-NMR (CDCl3, 300 MHz) δ (ppm): 3.65 (m, 24H, OCH2v–m), 3.79 (m, 4H, OCH2l), 4.03 (m, 4H, OCH2k), 4.62 (s, 4H, OCH2u), 5.41 (s, 2H, CH), 5.90 (m, 4H, pyr.), 6.12 (q, 4H, pyr., 3J = 3.0 Hz), 6.66 (m, 4H, pyr.), 6.77 (m, 6H, Ar), 7.18 (dd, 2H, Ar, 3J = 9.0 Hz, 3J = 7.5 Hz), 7.35 (d, 2H, Pyr, 3J = 7.5 Hz), 7.69 (t, 1H, Pys, 3J = 7.5 Hz), 8.15 (br, 4H, NH). 13C-NMR (CDCl3, 75 MHz) δ (ppm): 44.1, 67.4, 69.9, 70.4, 70.6, 70.8, 70.9, 107.2, 108.4, 112.9, 115.2, 117.3, 120.3, 121.2, 129.7, 132.5, 143.9, 157.7, 159.1.
Acknowledgements
We thank the University of Strasbourg, the International Centre for Frontier Research in Chemistry (FRC), Strasbourg, the Institut Universitaire de France (IUF), the Ministry of Education and Research and the CNRS for financial support. Thanks to Dr L. Allouche for NMR studies.Notes and references
- E. R. Kay, D. A. Leigh and F. Zerbetto, Angew. Chem., Int. Ed., 2007, 46, 72 CrossRef CAS.
- W. R. Browne and B. L. Feringa, Nat. Nanotechnol., 2006, 1, 25 CrossRef CAS.
- G. S. Kottas, L. I. Clarke, D. Horinek and J. Michl, Chem. Rev., 2005, 105, 1281 CrossRef CAS.
- G. Vives, H.-P. Jacquot de Rouville, A. Carella, J.-P. Launay and G. Rapenne, Chem. Soc. Rev., 2009, 38, 1551 RSC.
- J.-P. Sauvage, Molecular machines and motors, structure and bonding, Springer, Berlin, Heidelberg, 2001, vol. 99, pp. 1–282 Search PubMed; V. Balzani, M. Venturi and A. Credi, Molecular devices and machines: a journey into the nanoworld, Wiley-VCH, Weinheim, 2003, pp. 1–457 Search PubMed; T. R. Kelly, Molecular machines, Topics in Current Chemistry, Springer, Berlin, Heidelberg, 2005, vol. 262, pp. 1–227 Search PubMed.
- V. Balzani, A. Credi, F. M. Raymo and J. F. Stoddart, Angew. Chem., Int. Ed., 2000, 39, 3348 CrossRef CAS.
- K. Tashiro, K. Konishi and T. Aida, J. Am. Chem. Soc., 2000, 122, 7921 CrossRef CAS.
- M. Takeuchi, T. Imada and S. Shinkai, Angew. Chem., Int. Ed., 1998, 37, 2096 CrossRef CAS.
- (a) V. Balzani, M. Gomez-Lopez and J. F. Stoddart, Acc. Chem. Res., 1998, 31, 405 CrossRef CAS; (b) J. F. Stoddart, Acc. Chem. Res., 2001, 34, 410 CrossRef CAS; (c) V. Balzani, A. Credi, F. M. Raymo and J. F. Stoddart, Angew. Chem., Int. Ed., 2000, 39, 3348 CrossRef CAS; (d) A. H. Flood, R. J. A. Ramirez, W.-Q. Deng, R. P. Muller, W. A. Goddard III. and J. F. Stoddart, Aust. J. Chem., 2004, 57, 301 CrossRef CAS; (e) C. A. Schalley, K. Beizai and F. Vögtle, Acc. Chem. Res., 2001, 34, 465 CrossRef CAS; (f) A. M. Fuller, D. A. Leigh, P. J. Lusby, I. D. H. Oswald, S. Parsons and D. B. Walker, Angew. Chem., Int. Ed., 2004, 43, 3914 CrossRef CAS; (g) D. A. Leigh, P. J. Lusby, A. M. Z. Slawin and D. B. Walker, Angew. Chem., Int. Ed., 2005, 44, 4557 CrossRef CAS; (h) F. Coutrot and E. Busseron, Chem.–Eur. J., 2009, 15, 5186 CrossRef CAS; (i) E. Busseron, C. Romuald and F. Coutrot, Chem.–Eur. J., 2010, 16, 10062 CrossRef CAS.
- (a) J.-P. Sauvage, Science, 2001, 291, 2105 CrossRef CAS; (b) J.-P. Sauvage, Acc. Chem. Res., 1998, 31, 611 CrossRef CAS; (c) J.-P. Collin, C. Dietrich-Buchecker, P. Gavina, M. C. Jimenez-Molero and J.-P. Sauvage, Acc. Chem. Res., 2001, 34, 477 CrossRef CAS.
- (a) T. R. Kelly, H. De Silva and R. A. Silva, Nature, 1999, 401, 150 CrossRef CAS; (b) T. R. Kelly, Acc. Chem. Res., 2001, 34, 514 CrossRef CAS; (c) T. R. Kelly, X. Cai, F. Damkaci, S. B. Panicker, B. Tu, S. M. Bushell, I. Cornella, M. J. Piggott, R. Salives, M. Cavero, Y. Zhao and S. Jasmin, J. Am. Chem. Soc., 2007, 129, 376 CrossRef CAS.
- (a) N. Koumura, R. W. J. Zijlstra, R. A. van Delden, N. Harada and B. L. Feringa, Nature, 1999, 401, 152 CrossRef CAS; (b) M. K. J. ter Wiel, R. A. van Delden, A. Meetsma and B. L. Feringa, J. Am. Chem. Soc., 2003, 125, 15076 CrossRef CAS; (c) R. A. van Delden, M. K. J. ter Wiel, N. Koumura and B. L. Feringa, in Molecular Motors, ed. M. Schliwa, Wiley-VCH, Weinheim, 2003, pp. 559–577 Search PubMed.
- (a) J. F. Morin, Y. Shirai and J. M. Tour, Org. Lett., 2006, 8, 1713 CrossRef CAS; (b) Y. Shirai, J.-F. Morin, T. Sasaki, J. M. Guerrero and J. M. Tour, Chem. Soc. Rev., 2006, 35, 1043 RSC; (c) G. Vives and J. M. Tour, Acc. Chem. Res., 2009, 42, 473 CrossRef CAS.
- L. Grill, K.-H. Rieder, F. Moresco, G. Jimenez-Bueno, C. Wang, G. Rapenne and C. Joachim, Surf. Sci., 2005, 584, L153 CrossRef CAS.
- (a) A. Carella, J. Jaud, G. Rapenne and J.-P. Launay, Chem. Commun., 2003, 2434 RSC; (b) A. Carella, J.-P. Launay, R. Poteau and G. Rapenne, Chem.–Eur. J., 2008, 14, 8147 CrossRef CAS; (c) T. C. Bedard and J. S. Moore, J. Am. Chem. Soc., 1995, 117, 10662 CrossRef CAS; (d) K. Skopek, M. C. Hershberger and J. A. Gladysz, Coord. Chem. Rev., 2007, 251, 1723 CrossRef CAS; (e) E. B. Bauer, F. Hampel and J. A. Gladysz, Organometallics, 2003, 22, 5567 CrossRef CAS; (f) N. Weibel, A. Mishchenko, T. Wandlowski, M. Neuburger, Y. Leroux and M. Mayor, Eur. J. Org. Chem., 2009, 6140 CrossRef CAS.
- (a) A. Guenet, E. Graf, N. Kyritsakas, L. Allouche and M. W. Hosseini, Chem. Commun., 2007, 2935 RSC; (b) A. Guenet, E. Graf, N. Kyritsakas and M. W. Hosseini, Inorg. Chem., 2010, 49, 1872 CrossRef CAS.
- (a) T. Lang, A. Guenet, E. Graf, N. Kyritsakas and M. W. Hosseini, Chem. Commun., 2010, 46, 3508 RSC; (b) T. Lang, E. Graf, N. Kyritsakas and M. W. Hosseini, Dalton Trans., 2011, 40, 3517 RSC; (c) T. Lang, E. Graf, N. Kyritsakas and M. W. Hosseini, Dalton Trans., 2011, 40, 5244 RSC; (d) A. Guenet, E. Graf, N. Kyritsakas and M. W. Hosseini, Chem.–Eur. J., 2011, 17, 6443 CrossRef CAS.
- T. Lang, E. Graf, N. Kyritsakas and M. W. Hosseini, Chem.–Eur. J., 2012, 18, 10419 CrossRef CAS.
- (a) M. Momenteau and C. A. Reed, Chem. Rev., 1994, 94, 659 CrossRef CAS and references therein; (b) J. P. Collman, R. Boulatov, C. J. Sunderland and L. Fu, Chem. Rev., 2004, 104, 561 CrossRef CAS.
- (a) P. Leighton and J. K. M. Sanders, J. Chem. Soc., Chem. Commun., 1984, 854 RSC; (b) R. J. Abraham, P. Leighton and J. K. M. Sanders, J. Am. Chem. Soc., 1985, 107, 3472 CrossRef CAS.
- (a) J. Froidevaux, P. Ochsenbein, M. Bonin, K. Schenk, P. Maltese, J.-P. Gisselbrecht and J. Weiss, J. Am. Chem. Soc., 1997, 119, 12362 CrossRef CAS; (b) P. Ochsenbein, M. Bonin, K. Schenk, J. Froidevaux, J. Wytko, E. Graf and J. Weiss, Eur. J. Inorg. Chem., 1999, 1175 CrossRef CAS.
- P. D. Rao, S. Dhanalekshmi, B. J. Littler and J. S. Lindsey, J. Org. Chem., 2000, 65, 7323 CrossRef CAS; B. J. Littler, M. A. Miller, C.-H. Hung, R. W. Wagner, D. F. O'Shea, P. D. Boyle and J. S. Lindsey, J. Org. Chem., 1999, 64, 1391 CrossRef; D. Gryko and J. S. Lindsey, J. Org. Chem., 2000, 65, 2249 CrossRef.
- C. Acerete, J. M. Bueno, L. Campayo, P. Navarro and M. I. Rodriguez-Franco, Tetrahedron, 1994, 50, 4765 CrossRef CAS.
- B. Rezzonico and M. Grignon-Dubois, J. Chem. Res., 1995, 142 Search PubMed.
- Y. Nakamura, S. Takeuchi and Y. Ohgo, Tetrahedron, 1999, 55, 4595 CrossRef CAS.
- (a) S. Rucareanu, A. Schuwey and A. Gossauer, J. Am. Chem. Soc., 2006, 128, 3396 CrossRef CAS; (b) D. T. Gryko and M. Tasior, Tetrahedron Lett., 2003, 44, 3317 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef.
Footnotes |
| † This article is included in the All Aboard 2013 themed issue. |
| ‡ CCDC 894032 and 894033. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c2nj40657h |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2013 |