Properties of long alkyl-chained resorcin[4]arenes in bilayers and on the Langmuir trough†
Priyanka
Ogirala
a,
Saeedeh
Negin
a,
Ceno
Agena
b,
Christian
Schäfer
b,
Thomas
Geisler
b,
Jochen
Mattay
b and
George W.
Gokel
a
aCenter for Nanoscience, Departments of Chemistry & Biochemistry and Biology, University of Missouri – Saint Louis, Saint Louis, MO 63121, USA. E-mail: mattay@uni-bielefeld.de; gokelg@umsl.edu
bDepartment of Chemistry, Organic Chemistry I, Bielefeld University, P.O.Box 10 01 31, D-33501 Bielefeld, Germany
First published on 4th October 2012
Abstract
Two resorcinarenes and one cavitand having 10-(decylthio)decyl side chains were studied by using both a planar bilayer conductance voltage clamp device and a Langmuir trough. The former detects the presence of pore formation in a phospholipid bilayer and the latter probes the aggregation behavior of the amphiphiles as a monolayer. The Langmuir trough behavior of these amphiphiles showed that higher polarity head groups dominated organization, but as the head group polarity decreased, monolayer organization was dominated by the four side chains. Among the three, i.e., resorcinarene, octamethyl resorcinarene, and cavitand, only the methylated head group derivative formed ion-conducting pores in bilayers.
Introduction
The condensation of carbonyl compounds with electron-rich aromatic compounds has been known for more than a century. Von Baeyer condensed acetone with pyrrole to make compounds1 that are now called “calixpyrroles.”2 Baekelund developed the polymers known as Bakelite based on condensations of phenols with carbonyl compounds.3 With the advent of supramolecular chemistry, macrocycles such as those that Gutsche called calixarenes4 were extensively explored. At present, at least half a dozen monographs summarize the chemistry of calixarenes.4,5 Two families of macrocycles share a structural analogy with the calixarenes. They are resorcinarenes,6 which derive from resorcinol, and pyrogallolarenes, which are formed from pyrogallol.7 As expected in the supramolecular field, variants and hybrids have also emerged. These include, but are not limited to, calixcrowns,8 calix[4]resorcinarenes,9 and cavitands.10Amphiphiles that are based on the calixarene, resorcinarene, cavitand, and related scaffolds have shown activity both as carriers and as pore-formers that function in bilayer membranes. An early example is found in the functionalized calixarenes reported in 1985 by McKervey and collaborators11 to exhibit both ion binding and “ion transfer” properties. Carrier function was reported for molecules in this general class of compounds from the laboratories of Shinkai,12 Jin et al.,13 and Beer et al.14 Crown ether calixarene hybrids were reported in 1998 by de Mendoza et al.15 to transport sodium cations through phospholipid bilayers. A family of calixarene structures was reported in 2002 by Siderov, Davis, and their coworkers to form stable and functional pores in bilayers.16 Transport of water, i.e., aqua channel activity, by a self-assembled calixarene was reported by Coleman and coworkers.17 Other examples have been reported by Maulucci et al.,18 Davis et al.,19 and Prados, de Mendoza, et al.20 Cavitands having alkyl tails and amino acid “head groups” have recently been reported to show pore formation, the characteristics of which depend on the amino acid headgroup.21 The transport ability of certain of these systems has recently been reviewed.22
Pyrogallol[4]arenes (Pgs) are closely related to calixarenes and resorcinarenes. They are the acid-catalyzed condensation products of pyrogallol (1,2,3-trihydroxybenzene) and aldehydes. They form both bilayers and capsules23 in the solid state and the capsules may be further stabilized by replacing the hydrogen bond network with metal ions.24 We and others25 have explored the ability of amphiphilic pyrogallolarenes26 to form pores in bilayer membranes.27 These studies have been somewhat limited by the problem of purification. The condensation that produces the various macrocycles affords products having different sidechain stereochemistries. When all four chains are aligned on the same side of the macroring as shown for 1–3, the conformation is designated rccc to indicate that three chains are cis to a reference chain.28 When the sidechains are relatively long alkyl groups, crystallization is more difficult. In addition, variations in sidechain stereochemistry may complicate or prevent crystallization of any pure product. As a result, relatively long-chained examples of resorcinarenes and pyrogallolarenes are less well studied than their shorter-chained counterparts.
Studies of transport mediated in some way by resorcinarenes29 or amphiphilic cavitands have been reported.30 To our knowledge, the earliest example of an amphiphilic resorcinarene that formed ion-conducting pores was reported by Tanaka, Kobuke, and Sokabe.31 In this case, K+-selective pores were formed in bilayers by resorcinarenes having four n-heptadecyl tails. In previous work, we found that some pyrogallol[4]arenes27 and some cavitands21 having shorter tails formed pores that appeared to result from aggregation.
The work presented here had three key goals. First, we wished to compare compounds of three structural types that varied only in headgroup structure and polarity. To our knowledge, no such direct head group comparison has previously been reported. Second, the side chains present in these three related structures are quite long and oriented in the rccc fashion. To our knowledge, no comparable array of structures has been reported. Third, the three related structures would be assayed for their ability to form pores in phospholipid bilayers. Success was expected in this context based on the previous studies cited above.21,27
Results and discussion
Design rationale and compounds used in this study
Compound 1 was synthesized as previously reported by one of us.32 Compounds 2 and 3 were synthesized following a procedure described by Reinhoudt and coworkers.33 A single molecule atomic force microscopy study was also reported by one of us for compound 3.34 Each compound was characterized for purity by using NMR, mass spectrometry, and combustion analyses.The preparation of pyrogallol[4]arenes can be accomplished by heating an aldehyde and pyrogallol in ethanolic aqueous HCl. Although multiple chain orientations are possible, it is often the case that the Pg isomer having the four side chains parallel crystallizes from the solution. The chain orientation of many members of this resorcinarene and pyrogallolarene families has been confirmed by X-ray crystallography.35,36 As the lipid side chain length increases, it becomes less likely that the pure rccc isomer will crystallize from what is certainly a complex mixture. Thus, a key feature of this study was to use long lipid chains that have a known orientation. These were available from a previous study in which the chains were folded and the sulfur atoms served as anchors to a gold surface.34 In the present case, the use of sulfur as a linking element does not alter the hydrophobicity of the chain. The electronegativity of C and S are nearly identical (2.55 vs. 2.58), divalent sulfur is almost the same size as a methylene group, and the difference in bond angles is small. The sulfur was originally incorporated into the alkyl chains to provide an anchor point on a gold surface with the expectation that the chains would fold back, affording twice the molecular area of a single alkyl chain.37
The three compounds chosen for study all have four aromatic rings similarly linked and all have identical side chains. They differ in rigidity and in the oxygen functionality at the polar end. The differences are apparent in the structures of 1–3, shown in Fig. 1.
 | ||
| Fig. 1 Structures of compounds 1–4. | ||
Langmuir trough analyses of monolayers formed from 1–3
The Langmuir trough is a powerful tool to assess the behavior of amphiphiles that form monolayers on an aqueous surface.38 The amphiphile is dissolved in a water immiscible solvent and then deposited on a water surface in a trough. Evaporation of the solvent leaves the amphiphile distributed randomly on the water surface. The trough is equipped with movable barriers that compress the amphiphile. The change in surface pressure is monitored and plotted as a function of molecular area.Compounds 1–3 are amphiphiles, each of which contains more than 100 carbon atoms. All three compounds contain 8 oxygen atoms, but they are configured differently. The headgroup of 1 was expected to be more polar owing to its 8 hydroxyl groups compared either to the 8 methyl ethers or to the cavitand configuration. A simple log P calculation39 returned the following partition values: 1, 39.0; 2, 43.7; and 3, 44.0. The log P values for 2 and 3 are within error of each other, despite the fact that 3 has only half the number of carbons in its head groups compared to 2. Obviously, partition constants of this magnitude are approximate at best, but they confirm the expectation that 1 has a more polar head group than either 2 or 3. Moreover, since 2 and 3 have the same partition coefficients, differences in their behavior will likely result from differences in flexibility and oxygen position.
Fig. 2 shows the surface area-pressure isotherm for 1 when spread on highly purified water (see Experimental Section). The graph shows 9 nearly superimposable curves (different colors) that confirm the high reproducibility of the data. Five inflection points are apparent. We interpret these inflection points to have the following meaning. Organization begins to occur during the compression cycle at 335 Å2. The point at which an organizational transition to a higher order arrangement occurs is observed at 270 Å2. An inflection point is observed at 115 Å2, at which point the head groups start to interact directly. The transition continues to 105 Å2, which represents the closest packing organization on the water surface. From this final inflection point, the surface pressure rises rapidly as the amphiphiles begin to tumble over each other, perhaps forming a bilayer. The collapse point is shown at 25 Å2.
 | ||
| Fig. 2 Surface pressure–area (π–A) isotherm data for compound 1. Transitions observed are 335, 270, 115, 105 and 25 Å2. Each line represents the result of a separate experiment; the superimposition indicates the reproducibility. | ||
By using space filling Corey–Pauling–Koltun (CPK) molecular models, we estimated the diameter of the resorcinarene macrocycle to be ∼12 Å. This suggests a head group area for amphiphile 1 of ∼113 Å2. A transition in organization clearly occurs from 115–105 Å2. The measurement from physical models is in good agreement with the experimental range of 105 Å2, considering that variance of a single millimeter in the model measurement would result in ∼2 Å2 difference in area. We infer from the approximately 110 Å2 area range that compression of the monolayer leads to amphiphiles organized with the hydroxyl groups H-bonded to the aqueous phase and the hydrocarbon chains extended into the air phase. This conclusion is based in part on the area occupied by a hydrocarbon chain, namely ∼20 Å2. Four fully extended alkyl chains would occupy only about 80 Å2 rather than the observed ∼110 Å2. If the chains were folded over, the area occupied would be doubled, i.e., ∼160 Å2. We conclude that 1 forms a uniform, stable monolayer on the water surface and that its minimum area corresponds to the array of macrocycles in lateral contact with each other.
When the eight hydroxyl groups of 1 are methylated to form 2, the surface activity is altered. Simple chemical principles suggest that the macrocyclic head group of 2 will be less polar than that of 1 and space-filling molecular models suggest that it may occupy slightly more space owing to the difference in size between hydroxyl and methoxyl. Fig. 3 shows the results of seven Langmuir trough experiments conducted with 2 under conditions identical to those used for the analysis of 1.
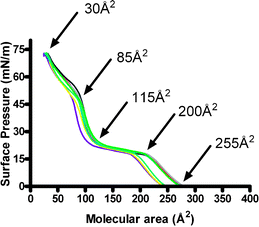 | ||
| Fig. 3 Surface pressure–area (π–A) isotherm data for compound 2. Transitions observed are 255, 200, 115, 85, and 30 Å2. Each line represents the result of a separate experiment; the superimposition indicates the reproducibility. | ||
Several differences are apparent in the isotherms observed for 1 and 2. Notably, the data for 2 (Fig. 3), while consistent, show somewhat more variation than those observed for 1 (cf. Fig. 2). The largest deviation is observed near a liquid expanded–liquid compressed inflection point from which little structural inference can be drawn. The data are consistent at the 115 Å2 inflection point that corresponds to our estimate of head group size, again as judged by measurements of CPK molecular models. It is at this point that the surface oriented amphiphiles begin to contact each other. The approximate surface pressure observed at this point is 20–25 mN m−1, which is similar to that observed for 1 at the corresponding point in Fig. 2. The surface pressure rose sharply for 1 from this point and collapse ensued. In the case of 2, a distinct inflection point is observed at 85 Å2. The surface pressure at this point is ∼50 mN m−1, suggesting a different and more stable organization of the amphiphiles. We infer this to be the interaction of the long alkyl chains that contact each other when the less polar head groups break linear contact with the water surface. The arrangement can be envisioned as a kind of molecular subduction, as we have previously observed for tetraisopropylpyrogallol[4]arene.40
As expected for doubly-linked arenes, CPK models showed that 3 has the least flexible headgroup of the three compounds studied. A comparison of the structures of 2 and 3 might lead one to conclude that the smaller number of carbon atoms in 3 would render it more amphiphilic. As noted above, the difference between 104 and 108 carbons does not substantially affect log P. Of course, the octahydroxylated compound (1) would remain the most amphiphilic compound among 1, 2, and 3. The methylene links that form the cavitand structure restrict the motion of the oxygen atoms and thus their accessibility to the water surface. As a result, the inflection point observed at ∼115 Å2 is absent in the isotherm illustrated in Fig. 4. Indeed, the transition from a gas phase or liquid expanded state to the assembly organized by alkyl chain contacts is quite clear. The surface pressure at the alkyl chain contact stage (∼80 Å2) is similar to that observed at the same stage for 2 in Fig. 3.
 | ||
| Fig. 4 Surface pressure–area (π–A) isotherm data for compound 3. Transitions observed are 150, 80, and 25 Å2. Each line represents the result of a separate experiment; the superimposition indicates the reproducibility. | ||
The behavior of 1–3 on the Langmuir trough can be summarized as follows. First, in none of the cases observed does the organization reflect the alkylthioether chains folding back on themselves to give the type of double chains for which they were originally designed. It should be noted that cavitand 3 does show an inflection point at ∼150 Å2, which suggests that the chains may initially be folded as compression moves from the liquid expanded surface state. Second, we infer that the stable monolayer formed by 1 involves a headgroup on the water surface and the chains unfurled and extended into the air phase. Compounds 2 and 3 have less polar headgroups and appear to condense until the extended alkylthioether chains are compressed to minimum area. This must force the relatively rigid headgroups to partly submerge in the aqueous phase.
The Brewster angle microscope permits the direct observation of the aqueous surface as pressure is applied by the moving barriers. No images were obtained from compression experiments involving either 1 or 2. A set of images was obtained for 3. The increasing organization of 3 on the trough surface is apparent in the Brewster angle microscopic (BAM) images shown in Fig. 5. Panels a–d show small segments of the trough in a surface area range of 251–182 Å2. An initial clustering of the cavitand is apparent in all four images, suggesting a tendency to aggregate despite the lack of confinement. The tendency of 3 to aggregate gives a non-uniform surface containing islands of material as shown in Fig. 5 and areas in which no material is apparent.
 | ||
| Fig. 5 Brewster angle micrographs for 3 captured at (a) 251 Å2, (b) 237 Å2, (c) 215 Å2, (d) 182 Å2, (e) 162 Å2, (f) 140 Å2. | ||
An inflection point was noted at ∼150 Å2 for 3 in the trough study illustrated in Fig. 4. Panel e in Fig. 5 shows a high level of organization at 162 Å2. This area corresponds to the volume of 8 alkyl chains, adding additional evidence that the alkyl chains in 3 initially fold as noted above. The initial folding and the resulting larger size of the molecular entity may cause the aggregation observed for 3 but not for 1 or 2. The image shown in panel f of Fig. 5 is the point beyond which no further surface variation is observed.
Planar bilayer conductance studies
Previous studies suggested that some pyrogallolarenes,27 resorcinarenes,31 and cavitands21 show ion-transporting membrane activity. Thus, compounds 1–3 were assayed for the ability to conduct ions by examining their behavior in a planar phospholipid (asolectin) bilayer membrane. The planar bilayer voltage clamp technique allows the direct observation of ion flux by permitting an amphiphile to insert in a membrane separating two buffer solutions. Evidence for pore formation is observed as current flow and so-called “open–close” behavior.41 In the present study, two aqueous KCl solutions buffered at pH 7 were separated by the bilayer membrane. Samples of 1–3 were dissolved either in trifluoroethanol or DMSO and then added to the buffer solution. Each amphiphile was added to only one of the buffer solutions (the cis chamber) and tested under a variety of conditions. No ion current could be detected at any applied potential between 30–70 mV for 1. Likewise, ion transport was not observed for compound 3, which proved to be nearly insoluble in the range of solvents tried. Thus, the lack of results for 3 may either reflect inactivity or poor solubility or both. In contrast, ion flux was observed for octamethoxyresorcinarene 2, although the pores formed were not well behaved.The lack of pore formation by 1 is both surprising and troubling. In principle, 1 differs little from the compound reported by Tanaka et al.31 to form stable pores (4, Fig. 1). It was noted in that report that no activity was observed when 4 was added on only one side of the membrane. However, pore formation was apparent when the ionophore was added on both sides of the bilayer, suggesting that C17-resorcinarenes were present in each leaflet of the membrane. Open–close behavior could then be accounted for by shifting of the two resorcinarenes within each leaflet of the bilayer relative to each other. The conductance observed for 4 was about 6 picoSiemens (pS) at an applied potential of −74 mV.
We find it difficult to account for the difference in behavior between 1 and 4. One possibility might be the membrane in which the studies were conducted. However, Tanaka et al. report their studies in “soybean lecithin bilayers” and our studies were done in soybean asolectin. In fact, these terms are synonyms. The previous and current studies are separated by some years, but the essential nature of the bilayers must be similar. A second possible difference is whether the alkyl chains in Tanaka's studies were in the rccc configuration. The authors assert that the “alkyl chains [of 4] are arranged in an all-axial, all-cis configuration…” The chains in 1 were understood to be in the rccc configuration based on earlier structural studies.42 This leaves the length of the chains and the presence of sulfur in each chain of 1 as the only differences.
The sidechains in the Tanaka et al. resorcinarene were –(CH2)16CH3 compared to –(CH2)10S(CH2)9CH3. The electronegativity value for C and S is essentially the same: ∼2.5. In the extended conformations, simple computational analyses show that n-heptadecane and di-n-decylsulfide have overall lengths of ∼20 Å and ∼25 Å, respectively. This compares to ∼12 Å for the n-undecyl side-chained pyrogallolarenes and cavitands previously studied. To place these numbers in perspective, it is generally accepted that the hydrophobic regime of a mammalian phospholipid bilayer is between 30–35 Å thick, depending on membrane composition.
The Tanaka et al. paper does not specify a pore size. In the model proposed by Tanaka et al., ions pass through the tetraaryl headgroup. The diameter of the opening within the resorcinarene macrocycle estimated by Corey–Pauling–Koltun (CPK) space-filling scale molecular models is 2.5–3 Å. Tanaka et al. offer experimental information about the conductance pore formed from 4. They note that the charge-selective pore favors K+ over Na+ by three-fold and that Rb+ blocks the pore. The respective unsolvated diameters of Na+, K+ and Rb+ are 1.98 Å, 2.74 Å, and 3.04 Å.43 Taken together, these observations suggest that when 4 was added to both sides of a bilayer, the pore that resulted had a diameter of slightly less than 3 Å.
Compound 2, in which all eight hydroxyl groups are methylated, showed some activity when added to one side of the membrane and clear evidence for controlled ion flux when added to both sides of the bilayer (Fig. 6). When added to both buffer solutions, open states of significant duration having a conductance of 40 picoSiemens (pS) were observed in 2 ([2] = 2 μM], applied potential 50 mV, Fig. 6a). The trace illustrated (each trace represents 3 replicates) in Fig. 6b shows the behavior of 2 when added at a concentration of 3 μM and an applied potential of 70 mV. The major open state had an amplitude of ∼10 picoAmps (pA) and a conductance of 140 pS. The value of 140 pS is more than 20-fold larger than the conductance state reported by Tanaka et al. for compound 4. Note that the ordinate in the graph of Fig. 6b (potential = 70 mV) is ten-fold that of panel 6a (potential = 50 mV) and that the time scales differ. Despite the formation of these large open states, the membranes remained intact.
![Planar bilayer conductance traces for compound 2. (a, top) [2] = 2 μM in both buffer solutions, applied potential = 50 mV. (b) [2] = 3 μM in both buffer solutions, applied potential = 70 mV.](/image/article/2013/NJ/c2nj40337d/c2nj40337d-f6.gif) | ||
| Fig. 6 Planar bilayer conductance traces for compound 2. (a, top) [2] = 2 μM in both buffer solutions, applied potential = 50 mV. (b) [2] = 3 μM in both buffer solutions, applied potential = 70 mV. | ||
We used the Hille equation44 to estimate the size of pores formed from 2 when applied to both sides of the bilayer (applied potential 50 mV, conductance 40 pS, diameter ∼4 Å and conductance 140 pS at 70 mV, ∼8 Å, Fig. 6). The Hille equation was devised to correlate conductance with such properties as resistivity, pore length, and diameter. It has most often been applied to protein channels and should be considered an approximation as used here. Based on an examination of molecular models, a pore ∼4 Å across could result from aggregation of 4 molecules of 2 and a pore of ∼8 Å could result from pentamer aggregation. It is not surprising that the aggregation states would differ at different applied potentials, but it is unclear why tetramer and pentamer states are favored over other possibilities in the particular environment.
Our work both with n-undecyl-sidechained pyrogallol[4]arenes27b and cavitands21 led us to conclude that the amphiphiles studied inserted only in the upper leaflet of the bilayer and that pores formed as a result of amphiphile aggregation. The “opening” in the lower leaflet was understood as a rearrangement of phospholipids as suggested for toroidal pore formation.45 We felt that the possibility of forming a dimeric pore based on oppositely aligned monomers was limited by the need for transverse relaxation or “flip–flop” by the amphiphiles.46 In those experiments, we added pyrogallol[4]arenes to only one side of the bilayer. In two previous studies, we examined pyrogallol[4]arenes having sidechains that ranged in length from methyl to undecyl27b and cavitands having undecyl side chains but varied head groups.21 Taken together, the evidence suggested that the ion flux recorded for these compounds resulted from aggregation of several monomers that created an opening within the bilayer. In all of the previously studied cases, the pyrogallol[4]arenes had shorter hydrocarbon chains than do compounds 1–3.
The data obtained in this case present a conundrum. While it is true that transmembrane conductance was observed when 2 was added to one side of the membrane, the results shown in Fig. 6 resulted when it was added to both cis and trans buffer chambers. If 2 forms pores that are aggregates having headgroups in only one leaflet of the bilayer, why are better results obtained when 2 is added to both leaflets? If the pores result from the type of lateral aggregation previously inferred, how do the aggregate pores gate?
It is known that membranes thin in proximity to pores. It is possible that aggregated pores form on each side of the bilayer and mediate ion flux. The extended chains of 2 are ∼25 Å long. Relatively little thinning of the insulator regime would be required for the decylthiodecyl chains to span it. This could account for aggregate function but, if correct, it is unclear why adding 2 to both sides of the bilayer is significantly better than adding to one side.
Correlation of ion flux and Langmuir trough data
The results of planar bilayer studies showed that 1 was inactive, 2 showed transient activity when added to one side of the bilayer but more robust pore formation when added to both sides. We can make no comment on the lack of membrane activity observed for 3 owing to its insolubility.The results for 1 were unexpected because 1 is quite similar to the Tanaka et al. compound (4). Since 1 has the most strongly anchoring head group, it may insert into the upper leaflet of the bilayer, as expected, but the pendant chains may favor the folded arrangement. In such a case, the combined area of the side chains (∼160 Å2) would exceed the headgroup area (∼110 Å2). The size of the alkyl chains could, on the one hand, prevent aggregation and organization of the resorcin[4]arenes in a fashion analogous to that inferred for pyrogallol[4]arenes.27 On the other hand, 1 would lack any opening through which ions could pass when inserted in the bilayer simply because the folded chains would block any orifice created by the headgroup. Data from the Langmuir trough confirm that the headgroup influence of 1 is greater than for either 2 or 3.
Octamethoxyresorcin[4]arene 2 presents the most interesting case. When added to both sides of the bilayer, an organized assembly was clearly formed. Based on the monolayer behavior observed on the Langmuir trough, we surmise that the side chains of 2 are extended on each side of the bilayer. Both the head groups and the side chains could have significant contact in the bilayer. Aggregation of 2 as 24 or 25 in a single bilayer leaflet accounts structurally for the pore sizes estimated from the conductance values. It is unclear, however, how aggregates in upper and lower leaflets would interact. It is also currently unclear why 1 is inactive, at least based on its similarity with literature precedent, and 2 shows versatile pore formation.
Conclusion
Compounds 1–3 have identical side chains but differ in the polar residues in the resorcinarene-type macrocyclic head groups. Compression led to a transition corresponding to the head group's size in 1 and 2. Compounds 2 and 3 compressed first to a head group area and then to an area corresponding to the extended side chains. Compound 1 apparently adhered to the aqueous phase until collapse with no corresponding transition. These studies showed clearly that in all cases, the decylthiodecyl chains were extended at maximum compression. Compound 3 showed a transition suggesting that its side chains were initially folded, but these unfurled as well leading from a 150 Å2 state to an 80 Å2 compressed state without an inflection point corresponding to head group contact. The difference in monolayer behavior for 3 was also evinced by Brewster angle microscopic images that showed considerable aggregation and that were not obtained either for 1 or 2. We conclude that in general, the long side chains in 1–3 control their assembly to an increasing extent as head group polarity decreases. The presence of the sulfur atom does not appear to play a major conformational role. Thus, for polar 1, head groups size dominates because the hydrophilic head group keeps the macrocycle firmly involved with the aqueous phase. In cavitand 3, the less polar head group is not as strongly bound to water and the side chains align as compression increases.Compound 3 was too insoluble to study by planar bilayer conductance. Surprisingly, resorcinarene 1 showed no evidence of pore formation when examined by this technique although previous literature reported activity for a close analog. Octamethoxyresorcinarene 2 showed some membrane activity when administered to one side of the bilayer, but reproducible activity when applied to both sides of the membrane. We conclude that aggregates of 2 form in each leaflet of the bilayer, but it is currently unclear how they interact to produce ion-conducting pores.
Experimental section
General information
The compounds used in this study were previously reported and are referenced in the text.Langmuir trough studies
HPLC grade CHCl3 (Aldrich) was used to prepare amphiphile solutions with a concentration of ∼1 mg mL−1 as determined by mass. Surface pressure–area isotherm experiments were carried out on a Langmuir trough (Nima, UK). Pressure was measured with a Wilhelmy plate made out of filter paper. Subphase temperature was maintained at 23.0 ± 0.1 °C by an Isotemp 3016 circulating thermostat. The subphase contained ultrapure water with a resistivity of 18.2 mΩ (Millipore). Monolayers were formed by spreading 50 mL of a CHCl3 solution of compounds 1–5 (1.0 mg mL−1) onto the subphase and allowing 10 minutes for the solvent to evaporate. Trough barriers were compressed at a constant speed of <0.3 nm2 mlc−1 min−1. Data were plotted as surface pressure (mN m−1) vs. molecular area (Å2). Isotherm data were collected in triplicate on each of 4 separate days, resulting in a total of 12 individual trials for each compound to obtain accurate isotherm information.Planar bilayer conductance studies
Planar bilayer conductance (also called BLM) experiments were performed by using a Warner BC-525D bilayer clamp apparatus. Planar membranes were formed by painting lipids (asolectin, 25 mg mL−1 in n-decane) over a 200 μm aperture on the side of a cuvette fitted into a chamber. The cuvettes contained a 450 mM KCl buffer solution (10 mM HEPES, pH = 7). After membrane formation was confirmed (capacitance > 100 pF), an aliquot of a trifluoroethanol or DMSO solution of the compound (concentration in the respective solvent = 1 mM) was stirred into the buffer in the cis chamber to achieve the desired concentration (typically 2–3 μM). Recordings were acquired with Clampex 9.2 (Axon Instruments) and data analyses were performed with Clamp fit 9.2 (Axon Instruments).Acknowledgements
JM thanks the Deutsche Forschungsgemeinschaft (SFB 613) for financial support. GWG thanks the NSF (CHE-0957535) for a grant that supported this work.Notes and References
- A. Baeyer, Ber. Dtsch. Chem. Ges., 1886, 19, 2184–2188 CrossRef.
- (a) P. A. Gale, J. L. Sessler, V. Král and Calixpyrroles, Chem. Commun., 1998, 1–8 RSC; (b) R. Custelcean, L. H. Delmau, B. A. Moyer, J. L. Sessler, W. S. Cho, D. Gross, G. W. Bates, S. J. Brooks, M. E. Light and P. A. Gale, Angew. Chem., Int. Ed. Engl., 2005, 44, 2537–2542 CrossRef CAS.
- (a) L. H. Baekeland, U. S. Patent, 942,699, Dec. 7, 1909 Search PubMed; (b) L. H. Baekeland, U S. Patent, 942,809, Dec. 7, 1909 Search PubMed.
- C. D. Gutsche, Calixarenes, Royal Society of Chemistry: Cambridge, 1989, vol. 1, p. 223 Search PubMed.
- (a) Calixarenes, a Versatile Class of Macrocyclic Compounds Topics in Inclusion Science, ed. J. Vicens and V. Böhmer, Springer Verlag, Berlin, 1990, p. 280 Search PubMed; (b) C. D. Gutsche, Calixarenes Revisited, Royal Society of Chemistry: Cambridge, 1998, vol. 6 Search PubMed; (c) Calixarenes in Action, ed. L. Mandolini and R. Ungaro, World Scientific Publishing Company, Hackensack, NJ, 2000, p. 271 Search PubMed; (d) C. D. Gutsche, Calixarenes: An Introduction (Monographs in Supramolecular Chemistry), Royal Society of Chemistry, Cambridge, UK, 2008, p. 248 Search PubMed; (e) Modern Supramolecular Chemistry: Strategies for Macrocycle Synthesis, ed. F. P. Diederich, P. J. Stang and R. R. Tykwinski, Wiley-VCH Verlag GmbH & Co., Weinheim, 2008, p. 400 Search PubMed.
- W. Sliwa and C. Kozlowski, Calixarenes and Resorcinarenes: Synthesis, Properties, and Applications, Wiley-VCH Verlag GmbH & Co., Weinheim, 2009 Search PubMed.
- S. J. Dalgarno, N. P. Power and J. L. Atwood, Coord. Chem. Rev., 2008, 252, 825–841 CrossRef CAS.
- L. Baldini, C. Bracchini, R. Cacciapaglia, A. Casnati, L. Mandolini and R. Ungaro, Chemistry, 2000, 6, 1322–1330 Search PubMed.
- V. K. Jain, S. G. Pillai, R. A. Pandya, Y. K. Agrawal and P. S. Shrivastav, Anal. Sci., 2005, 21, 129–135 CrossRef CAS.
- (a) D. J. Cram and D. J. Cram, Science, 1983, 219, 1177–1183 CrossRef CAS; (b) D. J. Cram, Cram, J. M. Container Molecules and Their Guests, Royal Society of Chemistry: Cambridge, 1994 Search PubMed; J. O. Green, J. H. Baird and B. C. Gibb, Org. Lett., 2000, 2, 3845–3848 Search PubMed.
- M. A. McKervey, E. M. Seward, G. Ferguson, B. Ruhl and S. J. Harris, J. Chem. Soc., Chem. Commun., 1985, 388–390 RSC.
- S. Shinkai, Bioorg. Chem. Front., 1990, 1, 161–195 Search PubMed.
- T. Jin, M. Kinjo, T. Koyama, Y. Kobayashi and H. Hirata, Langmuir, 1996, 12, 2684–2689 CrossRef CAS.
- P. D. Beer, P. A. Gale, Z. Chen, M. G. Drew, J. A. Heath, M. I. Ogden and H. R. Powell, Inorg. Chem., 1997, 36, 5880–5893 CrossRef CAS.
- J. Mendoza de, F. Cuevas, P. Prados, E. S. Meadows and G. W. Gokel, Angew. Chem., Int. Ed. Engl., 1998, 37, 1534–1537 CrossRef CAS.
- V. Sidorov, F. W. Kotch, G. Abdrakhmanova, R. Mizani, J. C. Fettinger and J. T. Davis, J. Am. Chem. Soc., 2002, 124, 2267–2278 CrossRef CAS.
- (a) A. W. Coleman, E. SilvaDa, F. Nouar, M. Nierlich and A. Navaza, Chem. Commun., 2003, 826–827 RSC; (b) A. Lazar, O. Danylyuk, K. Suwinska, F. Perret and A. W. Coleman, Chem. Commun., 2006, 903–905 RSC.
- N. Maulucci, F. Riccardis De, C. B. Botta, A. Casapullo, E. Cressina, M. Fregonese, P. Tecilla and I. Izzo, Chem. Commun., 2005, 1354–1356 RSC.
- (a) J. L. Seganish, J. C. Fettinger and J. T. Davis, Supramol. Chem., 2006, 18, 257–264 CrossRef CAS; (b) J. L. Seganish, P. V. Santacroce, K. J. Salimian, J. C. Fettinger, P. Zavalij and J. T. Davis, Angew. Chem., Int. Ed. Engl., 2006, 45, 3334–3338 CrossRef CAS; (c) O. A. Okunola, J. L. Seganish, K. J. Salimian, P. Y. Zavalij and J. T. Davis, Tetrahedron, 2007, 63, 10743–10750 CrossRef CAS.
- J. C. Iglesias-Sanchez, W. Wang, R. Ferdani, P. Prados, J. DeMendoza and G. W. Gokel, New J. Chem., 2008, 32, 878–890 RSC.
- I. Elidrisi, S. Negin, P. V. Bhatt, T. Govender, H. G. Kruger, G. W. Gokel and G. E. M. Maguire, Org. Biomol. Chem., 2011, 9, 4498–4506 RSC.
- G. W. Gokel and N. Barkey, New J. Chem., 2009, 33, 947–963 RSC.
- (a) T. Gerkensmeier, W. Iwanek, C. Agena, R. Fröhlich, S. Kotila, C. Näther and J. Mattay, Eur. J. Org. Chem., 1999, 2257–2262 CrossRef CAS; (b) T. Gerkensmeier, C. Agena, W. Iwanek, R. Frohlich, S. Kotilad, C. Nather and J. Mattay, Z. Naturforsch., 2001, 56b, 1063–1073 Search PubMed; (c) J. L. Atwood, L. J. Barbour and A. Jerga, Chem. Commun., 2001, 2376–2377 RSC.
- S. J. Dalgarno, N. P. Power, J. E. Warren and J. L. Atwood, Chem. Commun. (Cambridge, U. K.), 2008, 1539–1541 RSC.
- D. P. Nikolelis and S. S. Petropoulou, Biochim. Biophys. Acta, 2002, 1558, 238–245 Search PubMed.
- O. V. Kulikov, N. P. Rath, D. Zhou, I. A. Carasel and G. W. Gokel, New J. Chem., 2009, 33, 1563–1569 RSC.
- (a) O. V. Kulikov, R. Li and G. W. Gokel, Angew. Chem., Int. Ed. Engl., 2009, 48, 375–377 CrossRef CAS; (b) R. Li, O. V. Kulikov and G. W. Gokel, Chem. Commun., 2009, 6092–6094 RSC; (c) M. M. Daschbach, O. V. Kulikov, E. F. Long and G. W. Gokel, Chem.–Eur. J., 2011, 17, 8913–8921 Search PubMed; (d) S. Negin, M. M. Daschbach, O. V. Kulikov, N. Rath and G. W. Gokel, J. Am. Chem. Soc., 2011, 133, 3234–3237 CrossRef CAS.
- J. P. Kass, H. Cesar Zambrano, M. Zeller, A. D. Hunter and E. E. Dueno, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2006, E62, o3179–o3180 Search PubMed.
- L. Husaru, R. Schulze, G. Steiner, T. Wolff, W. D. Habicher and R. Salzer, Anal. Bioanal. Chem., 2005, 382, 1882–1888 CrossRef CAS.
- (a) V. K. Jain, S. G. Pillai, R. A. Pandya, Y. K. Agrawal and P. S. Shrivastav, Talanta, 2005, 65, 466–475 CrossRef CAS; (b) V. K. Jain, S. G. Pillai, R. A. Pandya, Y. K. Agrawal and P. S. Shrivastav, Anal. Sci., 2005, 21, 129–135 CrossRef CAS.
- Y. Tanaka, Y. Kobuke and M. Sokabe, Angew. Chem., Int. Ed. Engl., 1995, 34, 693–694 CrossRef CAS.
- P. Siffalovic, M. Michelswirth, P. Bartz, B. Decker, C. Agena, C. Schäfer, S. Molter, R. Ros, M. Bach, M. Neumann, D. Anselmetti, J. Mattay, U. Heinzmann and M. Drescher, J. Biotechnol., 2004, 112, 139–149 Search PubMed.
- E. Thoden van Velzen, J. F. J. Enbersen and D. N. Reinhoudt, Synthesis, 1995, 989–997 CrossRef.
- R. Eckel, R. Ros, B. Decker, C. Agena, J. Mattay and D. Anselmetti, Angew. Chem., Int. Ed. Engl., 2005, 44, 484–488 CrossRef CAS.
- G. W. V. Cave, S. J. Dalgarno, J. Antesberger, M. C. Farrarelli, R. M. McKinlay and J. L. Atwood, Supramol. Chem., 2008, 20, 157–159 CrossRef CAS.
- O. V. Kulikov, N. P. Rath, D. Zhou, I. A. Carasel and G. W. Gokel, New J. Chem., 2009, 33, 1563–1569 RSC.
- E. U. Thoden van Velzen, J. F. J. Engbersen, P. J. de Lange, J. W. G. Mahy and D. N. Reinhoudt, J. Am. Chem. Soc., 1995, 117, 6853–6862 CrossRef CAS.
- M. C. Petty, Langmuir-Blodgett Films: An Introduction, Cambridge University Press, Cambridge, 1996, p. 234 Search PubMed.
- ACD/Chemsketch Release 12.01, www.acdlabs.com.
- M. M. Daschbach, O. V. Kulikov, E. F. Long and G. W. Gokel, Chem.–Eur. J., 2011, 17, 8913–8921 Search PubMed.
- R. Ferdani and G. W. Gokel, Org. Biomol. Chem., 2006, 4, 3746–3750 RSC.
- Y. Aoyama, Y. Tanaka and S. Sugahara, J. Am. Chem. Soc., 1989, 111, 5397–5404 CrossRef CAS.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- B. Hille, Ionic Channels of Excitable Membranes, Sinauer Associates: Sunderland, MA, 3rd edn, 2001, pp. 9–352 Search PubMed.
- L. Yang, T. A. Harroun, T. M. Weiss, L. Ding and H. W. Huang, Biophys. J., 2001, 81, 1475–1485 CrossRef CAS.
- J. M. Berg, J. L. Tymoczko and L. Stryer, Biochemistry, W. H. Freeman, New York, 2012, p. 362 Search PubMed.
Footnote |
| † This article is included in the All Aboard 2013 themed issue. |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2013 |