One-step synthesis of monodisperse polydopamine-coated silver core–shell nanostructures for enhanced photocatalysis†
Jiu-Ju
Feng
*ab,
Pei-Pei
Zhang
b,
Ai-Jun
Wang
*ab,
Qi-Chen
Liao
b,
Jun-Lan
Xi
b and
Jian-Rong
Chen
a
aCollege of Chemistry and Life Science, College of Geography and Environmental Science, Zhejiang Normal University, Jinhua 321004, China. E-mail: jjfengnju@gmail.com; ajwangnju@gmail.com
bKey Laboratory of Green Chemical Media and Reactions, Ministry of Education, School of Chemical and Environmental Science, Henan Normal University, Xinxiang, Henan 453007, China
First published on 15th November 2011
Abstract
Nanosized silver (Ag) and polydopamine (PDA) composite was prepared via one-step polymerization of dopamine, using Ag+ ion as an oxidant. The oxidization of dopamine and the reduction of Ag+ ion occurred simultaneously in one-step, which resulted in a well-defined core–shell structure of PDA coating around the Ag nanoparticles (AgNPs). Its morphology, structure, and composition were characterized by scanning electron microscopy (SEM), transmission electron microscopy (TEM), X-ray power diffraction (XRD), UV-vis, Fourier transform infrared (FT-IR) spectroscopy, cyclic voltammograms (CVs) and electrochemical impedance spectroscopy (EIS), respectively. The composite showed good performance toward the photo-catalytic degradation of neutral red under UV irradiation. The possible mechanism regarding the plasmon-induced photocatalysis was discussed in some detail, where the main active species of O2˙− on the surface of the AgNPs were involved in this photoreaction system. The PDA shells greatly improve the photocatalysis due to the existence of the π–π* electron transition under UV light.
Introduction
Silver nanoparticles (AgNPs) have wide applications, including photography, catalysis, optics, and Raman spectroscopic applications.1–7 The main disadvantages of the silver structures are poor stability and narrow potential window because of its low oxidation potential. Therefore, coating silver nano-/micro-structures with a thin dielectric layer such as thiols is usually adopted to have better and broader applications in photo-electrochemistry.8,9 In our previous work, silver nanocorals were grown on graphite electrodes10 and subsequently coated with mercaptohexanoic acid by self-assembly. The presence of the organic layer is good to improve its stability and biocompatibility, due to the isolation of the silver surface from the aqueous solution. A similar strategy is suitable for the silver pillar-like arrays,1 which were subsequently coated with cysteamine as an isolated layer.With the tremendous advance of nanoscience and nanotechnology, silver involved heterostructures or composites have gained great interest for their enhanced photo/electro-catalytic activity and significant practical values.11–13 For example, Ag/TiO2-nanotubes were synthesized and displayed higher photocatalytic activity than bare TiO2-nanotubes toward the degradation of Rhodamine-B.14 Similar observation was observed on the dimer-type heterostructure of Ag–ZnO nanofibers and found that the heterojunction structure significantly enhanced the photocatalytic efficiency by the degradation of the same dye under UV irradiation.4 Recently, silver–polymer composites, especially conducting polymers,15 have attracted more attention in catalysis, optical and electrical fields,16–20 for their integrated properties and synergistic effects of the individuals. For example, silver@polypyrrole core–shell nanosnakes were synthesized by one-step hydrothermal treatment.21 In another example, AgNPs–polyaniline composites were obtained through a two-phase water/toluene interfacial reaction, which presented very specific electric behaviors.22
Self-polymerization of dopamine could be easily carried out on different substrates.23 The formed polydopamine (PDA) films have good conductivity, biocompatibility, and stability. Furthermore, it could act as a versatile platform for secondary surface-mediated reactions, even selective reactions, leading to tailoring of coatings for diverse uses.24,25 Recent investigations have demonstrated that the formed PDA films can be easily used in bioelectronic devices.26 For instance, PDA–enzyme–metallic nanoparticles (Au or Pt) hybrid films were developed for the fabrication of glucose and/or galactose biosensor based on the immobilized glucose oxidase or galactose oxidase.27
Previously, researchers have deposited the PDA films around the colloidal particles such as SiO228 and polystyrene29 spheres. In their cases, preparation of the colloidal particles and subsequent coatings usually involve two steps. Nonetheless, in the present work, we demonstrate a novel method for one-step synthesis of monodisperse PDA-coated silver nanoparticles (AgNPs@PDA) with well-defined core–shell structures.
Organic dyes are a significant part of the textile industries waste. When the textile dyes are released into water bodies, they can be toxic to the marine plants and species.30 Therefore, it is important to treat the organic dye wastes prior to discharging them into the environment. Photocatalytic treatment of these dyes using solar radiation using a proper photocatalyst is widely adopted, since it can assist in the degradation of the dyes to benign by-products and also help in the minimization of the number of steps required in textile effluent treatment.31,32
Here, we report a simple, one-step synthesis of core–shell AgNPs@PDA structures via simple oxidative polymerization of the DA by Ag+ ion, where no additional reagents or treatment are involved. The DA serves as a reducing agent and a monomer, while Ag+ ion as the oxidant to trigger the DA polymerization and the source of the metallic nanoparticles. As a result, the PDA film formed around the AgNPs outside. This simple approach provides a new strategy for the size-controlled synthesis of the AgNPs@PDA composites, which revealed high photocatalysis on the model dye of neutral red (NR).
Experimental
Reagents and apparatus
AgNO3, NR, DA, and other reagents were obtained from Shanghai Chemical Reagent Co. Ltd. (Shanghai, China), which were used without further purification. Double distilled water was used for preparing all of the solutions. DA solution was freshly prepared in 10 mM carbonate/bicarbonate buffer (pH 8.5). Phosphate solution (20 mM) was prepared by mixing the stock solutions of NaH2PO4 and Na2HPO4 together.Electrochemical experiments were performed on a CHI 660C electrochemical workstation (Shanghai Chenhua, China). All experiments were performed using a conventional three-electrode system, where the modified glassy carbon electrode (GCE) was used as the working electrode, a platinum wire as the counter electrode, and a saturated calomel electrode (SCE) as the reference electrode. The electrochemical impedance spectroscopy (EIS) was performed in 0.1 M KNO3 aqueous solution containing 5.0 mM Fe(CN)63−/Fe(CN)64− (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), using an alternating current voltage of 5.0 mV. The impedance measurements were conducted at an open circuit potential of 180 mV within the frequency range of 10−2–105 Hz. All experiments were performed at room temperature.
:1), using an alternating current voltage of 5.0 mV. The impedance measurements were conducted at an open circuit potential of 180 mV within the frequency range of 10−2–105 Hz. All experiments were performed at room temperature.
The morphologies of the bare PDA and AgNPs@PDA structures were investigated by scanning electron microscopy (SEM, JEOL/EO, Japan) and transmission electron microscopy (TEM, Hitachi, H-7500). UV-Vis and FT-IR spectra were recorded on a UV-2201 spectrophotometer (Shimadzu, Kyoto, Japan). FI-IR measurements were carried out at room temperature on a Nicolet Nexus 670 FT-IR spectrometer (America). The crystal structure of the prepared products was analyzed by X-ray diffraction (XRD). The patterns were recorded by using a Bruker-D8-AXS diffractometer system equipped with Cu Kα radiation (Bruker Co., Germany). Raman spectra measurements were carried out with excitation laser of Ar+ 514 nm (Innova 300, Coherent) using a confocal Raman microscope (LabRam HR-800, Jobin Yvon) equipped with a N2(l)-cooled back-illuminated charge-coupled device (CCD) detector.
Construction of AgNPs@PDA composites
Under stirring, 5 mL AgNO3 solution (5 mM) was quickly added into 10 mL freshly prepared DA solution (5 mM, dissolved in 10 mM carbonate/bicarbonate solution with pH 8.5), and reacted for 5 min. The products were collected by centrifugation, thoroughly rinsed with water, and dried at 60 °C under vacuum. For comparison, single PDA was also prepared without AgNO3 present in the reacted system under the similar conditions, with a reaction time of at least 30 min.Synthesis of AgNPs
AgNPs were prepared by aqueous reduction of AgNO3 with trisodium citrate following the method of Wang et al.33 Specifically, 18 mg silver nitrate was suspended in 100 mL water at 45 °C and rapidly heated to boiling before 2 mL trisodium citrate solution (1%) was added under vigorous stirring. The solution was held at boiling for 90 min and continuously stirred upon cooling. The precipitates were centrifuged and completely washed with ethanol and water, respectively. Finally, the products were dried in vacuum at 60 °C overnight for further characterization. The TEM image of the AgNPs is shown in Fig. S1 (ESI†).Fabrication of the modified electrodes
The glassy carbon electrodes (GCE) were polished with 1.0, 0.3, and 0.05 μm alumina slurry, followed by completely rinsing with water and drying at room temperature. The obtained samples of 10 mg AgNPs@PDA, PDA powder or AgNPs were re-dispersed in 2.0 mL water and ultra-sonicated for 5 min. Then, 10 μL of the 5 mg·mL−1 colloidal solutions were dropped onto the clean GCE surface and allowed to dry naturally.Photocatalytic degradation studies
NR, a kind of azine dye, was used as a model contaminant to test the photocatalytic behavior of the as-prepared samples.34,35 The comparison of the photocatalysis and degradation efficiency with previously studies is listed in Table S1 (ESI†). Photocatalytic experiments were performed under illumination using two commercially available 15 W Philips F15T8 black-light fluorescent bulbs (model 392233) that have spectral energy distribution centered at 254 nm. The intensity of the radiation reaching the solution surface (3.5 mJ·cm−2) was detected using a Chromaline UV Minder radiometer (UVM 226) connected to a remote probe (UVM 226 S). The UV light was provided by a 250 W high-pressure fluorescent Hg lamp (Institute of Electrical Light Source, Beijing; the strongest emission at 254 nm). It has been documented that much of the irradiation occurs within a few centimetres of the liquid surface (even at very low catalyst loadings).36,37 Here, 3 mL NR aqueous solution (50 μM) was used for the degradation. Typically, 1.83 mg of the sample of AgNPs@PDA was added into 3 mL NR solution, and irradiated under UV light for 3 min, and the peak absorbance was analyzed using a TU-1810 UV-Vis spectrophotometer (Beijing Purkinje General Instrument Co., Ltd.). Control experiments were performed without and with single PDA (or AgNPs) in bare NR solution by conducting UV irradiation. To ensure good repeatability of the absorption measurements, each experiment was conducted in the dark at least three times.Results and discussion
Morphology of the AgNPs@PDA nanostructures
The morphologies of AgNPs@PDA and PDA products were characterized through SEM and TEM experiments, respectively (Fig. 1). For the AgNPs@PDA, well-defined core–shell nanostructures are observed by TEM (Fig. 1A) and SEM images (Fig. 1C), respectively. Evidently, the dark core with the size of ca. 100 nm is assigned to the one-pot formed AgNPs for their high electron cloudy density. Meanwhile, a surrounding gray sheath about 10 nm in thickness is ascribed to the PDA coating. The contrast between the dark inner core and light sheath coating around can be easily distinguished. Here, DA simultaneously served as a reducer and a source of polymer, while Ag+ ion as an oxidant and a source of AgNPs at the same time. Thus, the preparation of AgNPs@PDA was very simple and fast, which can be completed within 5 min with a yield of 85%. Additionally, the experiments were easily repeated, which showed good reproducibility. This point is very crucial for the generation of nanostructures in nanotechnology. | ||
| Fig. 1 TEM (A, B) and SEM (C, D) images of the synthesized AgNPs@PDA (A, C) and pure PDA (B, D) structures. | ||
For comparison, TEM (Fig. 1B) and SEM (Fig. 1D) images of the bare PDA were also recorded. There are no dark inner cores observed across the whole section, just some gray particles are decorated. The light electron density is similar to that of the PDA coating on the AgNPs@PDA composites, which can be ascribed to the PDA. The TEM images further confirm the probable deposition of the PDA polymer at the surface of the Ag nanoparticles.
The bare PDA nanostructures can be formed by the DA self-polymerization in the presence of oxygen in solution. The whole reaction time was about 30 min, much longer than that using Ag+ ion as the oxidative reagent in our study, but similar to that observed by other groups.38 Taken all together, we can conclude that the DA self-polymerization prefers to take place on the surface of the AgNPs rather than in solution, and thereby resulting in the formation of the well-defined AgNPs@PDA core–shell structures, due to high affinity between the AgNPs and PDA.
In order to provide more detailed structural information about the products, we investigated the HRTEM and Raman spectra analysis (Fig. 2). As shown in Fig. 2A, Both AgNPs core and PDA shell are clearly observed in the HRTEM image. Moreover, nearly no signal is detected for bare AgNPs (Fig. 2B, curve a) or PDA samples (Fig. 2B, curve b). However, strong peaks are observed at 1368 cm−1 and 1603 cm−1 for the sample of AgNPs@PDA (Fig. 2B, curve c), which result from the stretching and deformation of aromatic rings of the PDA.39 All the results reveal the synthesis of the core–shell AgNPs@PDA composites.
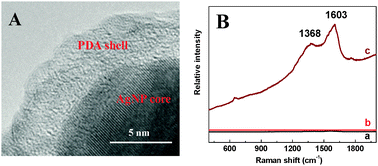 | ||
| Fig. 2 (A) Typical HRTEM image of the AgNPs@PDA. (B) Raman spectra of the AgNPs (a), PDA (b) and AgNPs@PDA (c). | ||
The XRD patterns of the PDA, AgNPs, and AgNPs@PDA are shown in Fig. 3. For both AgNPs (Fig. 3b) and AgNPs@PDA (Fig. 3c) samples, four characteristic peaks of silver (2θ = 38.08°, 44.42°, 64.40° and 77.84°) are clearly observed, corresponding to the (111), (200), (220) and (311), respectively. These results match well with the database of the face-centered cubic crystal of silver (JCPDS No. 65-2871).40 Additionally, for the sample of AgNPs@PDA, there is an extra diffraction peak appeared in the range of 20°–30°, similar to that of the bare PDA (Fig. 3a). Therefore, the broad peak is ascribed to the amorphous crystallinity of the PDA. All the results reveal the successful synthesis of the core–shell AgNPs@PDA composites.
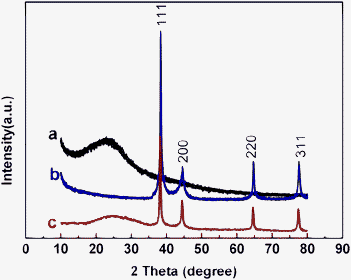 | ||
| Fig. 3 XRD patterns of the PDA (a), AgNPs (b), and AgNPs@PDA (c). | ||
The adsorption peak of the DA (Fig. 4A, curve a) and Ag+ ion (Fig. 4A, curve b) appeared at 280 nm and 270 nm, respectively. Both peaks almost disappeared after the DA and AgNO3 solutions were mixed together, since Ag+ ion oxidized DA to form PDA. Meanwhile, the Ag+ ion was reduced to metallic Ag. For the AgNPs@PDA sample, the typical peak is observed at 425 nm (Fig. 4A, curve d), which is due to the formation of the AgNPs,33 in agreement with that of the bare AgNPs (data not shown). Furthermore, there are two ultra-weak peaks appearing at 218 nm and 283 nm, similar to that observed for the PDA sample (Fig. 4A, curve c), which are originated from the π–π* electron transition of the PDA. These results further indicate that the AgNPs exist in the AgNPs@PDA composites.
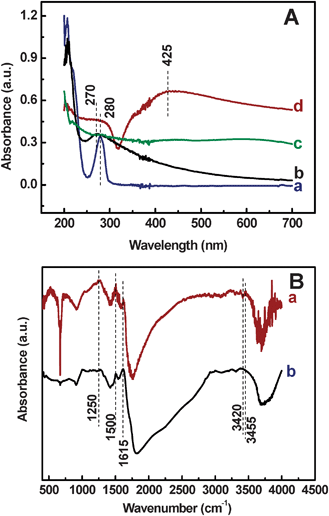 | ||
| Fig. 4 (A) UV-vis spectra of the DA (a), Ag+ (b), PDA (c) and AgNPs@PDA (d). (B) FT-IR spectra of the AgNPs@PDA (a) and PDA (b). | ||
The FT-IR spectra of the AgNPs@PDA sample display significant difference from the bare PDA sample. For the AgNPs@PDA sample, a typical band at 3455 cm−1 is due to the catechol hydroxyl groups mode in PDA coating (Fig. 4B, curve a), indicating the formation of PDA coatings on AgNPs. Similar observation is observed for the bare PDA sample, while the band negative shifts to 3420 cm−1 (Fig. 4B, curve b), in agreement with that reported by Fei et al.41 The peak intensities at 690 cm−1, 950 cm−1, 3455 cm−1, and 3750 cm−1 are different from those of the pure PDA sample, which might be ascribed to the effects of the AgNPs. These results further demonstrate the synthesis of the PDA shells around the AgNPs.
Electrochemical performance of the modified electrode
Cyclic voltammograms of Ru(NH3)6Cl3 are valuable and convenient tools for monitoring the surface status and the barrier of the modified electrode, because the electron transfer between the solution species and the electrode must occur by tunneling either through the barrier or through the defects in the barrier.42 Therefore, Ru(NH3)6Cl3 was chosen as a probe to monitor the whole process for the construction of the AgNPs@PDA modified electrode (Fig. 5A). The modification of different materials on the GCE is accompanied by a change in the amperometric response and a variation of the peak to peak separation between the cathodic and anodic peaks (ΔEp) of the redox probe. A quasi-reversible one-electron redox behavior of the probe is observed on the bare GCE (Fig. 5A, curve a) with a ΔEp of 96 mV. However, at the PDA modified electrode, an obvious increase in cathodic and anodic peak currents is observed, accompanied by the enlarged ΔEp (Fig. 5A, curve c), due to the increase of the real surface area. While, at the AgNPs modified GCE (Fig. 5A, curve b), besides the redox peaks of Ru(NH3)6Cl3, there is another typical couple of redox peaks of AgNPs (0.157 V, −0.135 V). Furthermore, at the AgNPs@PDA modified GCE (Fig. 5A, curve d), the currents of the redox peaks greatly increase and the ΔEp is decreased. Moreover, the typical peak is observed at 0.154 V and a weak peak could be seen at −0.120 V, assigned to the oxidation and reduction peaks of the AgNPs, respectively.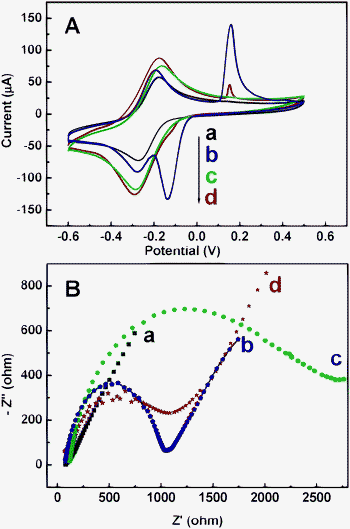 | ||
| Fig. 5 (A) Cyclic voltammograms of the bare (a), AgNPs (b), PDA (c) and AgNPs@PDA (d) modified electrodes in 5.0 mM Ru(NH3)6Cl3 at 0.1 V s−1. (B) Electrochemical impedance spectra (EIS) of the bare (a), AgNPs (b), PDA (c) and AgNPs@PDA (d) modified electrodes in 0.1 M KNO3 solution containing 10 mM Fe(CN)63−/Fe(CN)64− (1:1). | ||
Different sample modified electrodes were also recorded in phosphate solution (Fig. S2, ESI†). These results further revealed the formation of AgNPs in the composites.
As known, electrochemical impedance spectroscopy (EIS) is an effective tool for studying the interface properties of different materials modified electrodes. The impedance spectrum includes a semicircle portion at high frequencies corresponding to the electron transfer limited process, and a linear part at the low frequencies resulting from the diffusion limited electrochemical process.43 The semicircle diameter of the EIS equals to the electron transfer resistance (Ret), which controls the electron transfer kinetics of the redox probe (K4[Fe(CN)6]/K3[Fe(CN)6]) at the interface.44 Hence, the change in its value characterizes the modification of the electrode surface. As displayed in Fig. 5B, the assembly of the PDA film on the electrode increases the Ret (Fig. 4B, curve c), much larger than those of the AgNPs modified electrode (Fig. 5B, curve b) and bare electrode (Fig. 5B, curve a). The reason is that the PDA blocked the electron transfer of the probe. However, the AgNPs@PDA modified electrode (Fig. 5B, curve d) shows a smaller Ret. The AgNPs in PDA structures greatly facilitate the electron transfer between the probe and the electrode, owing to its own good conductivity. The results are similar to those obtained from cyclic voltammetry experiments.
Photocatalytic degradation studies
In order to assess the photocatalytic performance of the AgNPs@PDA for application in chemical oxidative processes, consideration of the photocatalysis process is necessary. In this study, NR is selected as a representative dye to evaluate the photocatalytic performance of the AgNPs@PDA composites. The decolorization of NR is monitored by UV-vis experiments. The time-dependent UV-vis spectra of the dye solution are shown in Fig. 6A. When 1.83 mg AgNPs@PDA powder was dispersed into 50 μM NR solution, and irradiated by UV light (λ = 254 nm) for a certain time, the absorbance peaks at 273 and 527 nm are simultaneously decreased to a large degree, respectively. After 5 min irradiation, the absorbance peak intensities of the NR solution are obviously decreased (Fig. 6B, curve d) with the reaction rate constant of 0.0956 min−1. As time elapsed, the intensities are steeply reduced with the rate constant of 0.0056 min−1. Conversely, when the initial NR solution was treated without any catalyst (Fig. 6B, curve a) or with bare PDA (Fig. 6B, curve b) in a similar way, negligible decolorization of the dye with the rate constants of 0.0021 min−1 and 0.0036 min−1 is observed under UV light for 50 min, respectively. Furthermore, when using the bare AgNPs as the catalyst, there are also two stages. Nonetheless, the intensities of the absorbance peaks decrease slowly, compared with those of the AgNPs@PDA. The corresponding rate constants are smaller than those of the AgNPs@PDA. That is, the photocatalytic ability of the AgNPs@PDA is stronger than that of the bare AgNPs. This is ascribed to the synergistic effect of the Ag core and PDA shell, which improve the photocatalytic capability because of the existence of the π–π* electron transition.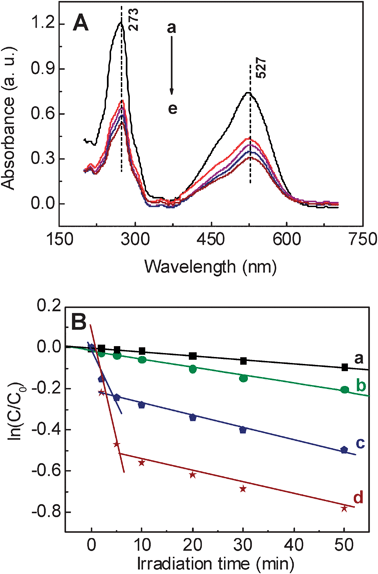 | ||
| Fig. 6 (A) UV-vis spectra from the 3 mL, 50 μM NR solution with 1.83 mg AgNPs@PDA at different time intervals (a–e: 0 min, 10 min, 20 min, 30 min and 50 min). (B) The logarithm of the ratio between the initial dye concentration and the one after photocatalytic degradation by pure PDA (b), AgNPs (c), AgNPs@PDA (d), and without any catalyst (a) (ln(C/C0)) vs. the corresponding irradiation time (min). | ||
Based on these results, we can conclude that a plasmon-induced mechanism of photocatalysis32,45–47 plays an important role during the photocatalytic degradation process. Specifically, the plasmon-induced production of superoxide radicals and excited H+ on the surface of the AgNPs@PDA are the main active species. Firstly, AgNPs can photo-induce the formation of electrons and holes with the irradiation of UV light. Then, the holes can be trapped by hydroxyl on the surface or in bulk solution, resulting in the formation of hydroxyl radical species (˙OH). Meanwhile, the photogenerated electrons can be captured by the adsorbed molecular oxygen (O2) and then produced superoxide radicals (O2˙−). The formed superoxide radicals are able to oxidize the dyes, such as NR in our system, due to their high oxidative activity, and thereby increase the degradation kinetics.48 The photocatalytic process of the AgNPs@PDA composites for the degradation of NR can be proposed as follows:
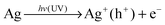
| e− + O2 → O2˙− |
| NR + Ag+ (h+) → Degradation products + Ag |
| NR + O2˙− + H2O → Degradation products + 4OH− |
The electron in the conduction and valence bands from plasmon-induced AgNPs could reduce oxygen to superoxide radicals, a similar phenomenon is observed on the AgI covered Ag electrode.49 The generated superoxide radicals migrated from inner AgNPs to the PDA shells outside. The PDA shell, because of its special structures and property, can improve the catalyst. Firstly, the PDA shell can adsorb NR onto the surface. Secondly, the PDA shell has the ability to photo-induce the formation of holes under UV light, which can prolong the recombination rate of photo-induced electrons and holes. As a consequence, the lifetime of the electron pairs is increased, due to the existence of the π–π* electron transition under UV light (Fig. 7). Therefore, the cooperation of the AgNPs and PDA shells greatly improves the photocatalytic efficiency of the NR degradation in this work.
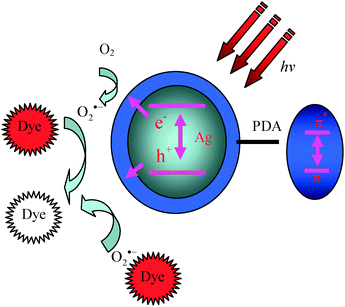 | ||
| Fig. 7 Schematic illustration of the photocatalysis mechanism of AgNPs@PDA. | ||
For comparison, using bare PDA as a catalyst, the photocatalytic degradation experiments were performed under the identical conditions. Obviously, the peak intensities of the NR are slightly decreased (Fig. 6B, curve b). In addition, negligible decolorization (<8.7%) of the NR self-photodegradation was observed similarly (Fig. 6B, curve c). These results confirm that the AgNPs@PDA nanostructures display good performance toward the photocatalytic degradation of NR.
In order to investigate the effect of UV-vis irradiation on NR degradation, control experiments are performed. That is, 1.83 mg AgNPs@PDA composite was put into 50 μM NR solution, without irradiation with UV light (Fig. 8). As expected, without irradiation (Fig. 8, curve b), the AgNPs@PDA composites exhibit much lower photocatalytic activity, compared to that under the irradiation of UV light (Fig. 8, curve a).
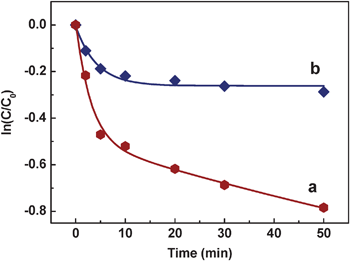 | ||
| Fig. 8 The logarithm of the ratios between the initial concentration of the dye and the ones after photocatalytic degradation by the AgNPs@PDA (ln(C/C0)) vs. the corresponding irradiation time in the presence (a) or absence (b) of UV light. | ||
Conclusions
We develop a simple wet-chemical method for the rapid synthesis of monodisperse, core–shell AgNPs@PDA nanostructures. The present study outlines the use of DA for efficient reduction of Ag+ ion to form AgNPs–PDA core–shell nanostructures. Because of the easy preparation, low cost and high photocatalytic ability of the hybrid composites, the present study enlarges the application to degrade other organic pollutants.Acknowledgements
We greatly acknowledge the financial support of the Natural Science foundation of China (No. 20805011, 20905021, 21175118).References
- J.-J. Feng, Y.-H. Lu, U. Gernert, P. Hildebrandt and D. H. Murgida, J. Phys. Chem. C, 2010, 114, 7280 CAS.
- G. Wang, W. Wang, J. Wu, H. Liu, S. Jiao and B. Fang, Microchim. Acta, 2009, 164, 149 CrossRef CAS.
- W. Ngeontae, W. Janrungroatsakul, P. Maneewattanapinyo, S. Ekgasit, W. Aeungmaitrepirom and T. Tuntulani, Sens. Actuators, B, 2009, 137, 320 CrossRef.
- D. Lin, H. Wu, R. Zhang and W. Pan, Chem. Mater., 2009, 21, 3479 CrossRef CAS.
- O. Akhavan, M. Abdolahad, Y. Abdi and S. Mohajerzadeh, J. Mater. Chem., 2011, 21, 387 RSC.
- M. Potara, A.-M. Gabudean and S. Astilean, J. Mater. Chem., 2011, 21, 3625 RSC.
- W.-P. Xu, L.-C. Zhang, J.-P. Li, Y. Lu, H.-H. Li, Y.-N. Ma, W.-D. Wang and S.-H. Yu, J. Mater. Chem., 2011, 21, 4593 RSC.
- B. Yin, H. Ma, S. Wang and S. Chen, J. Phys. Chem. B, 2003, 107, 8898 CrossRef CAS.
- S. Fujii, A. Aichi, K. Akamatsu, H. Nawafune and Y. Nakamura, J. Mater. Chem., 2007, 17, 3777 RSC.
- J.-J. Feng, P. Hildebrandt and D. H. Murgida, Langmuir, 2008, 24, 1583 CrossRef CAS.
- M. Es-Souni, H. Fischer-Brandies and M. E-Souni, Adv. Funct. Mater., 2008, 18, 3179 CrossRef CAS.
- D. D. Evanoff and G. Chumanov, ChemPhysChem, 2005, 6, 1221 CrossRef CAS.
- V. Biju, T. Itoh, A. Anas, A. Sujith and M. Ishikawa, Anal. Bioanal. Chem., 2008, 391, 2469 CrossRef CAS.
- G. Guo, B. Yu, P. Yu and X. Chen, Talanta, 2009, 79, 570 CrossRef CAS.
- G. Nesher, M. Aylien, G. Sandaki, D. Avnir and G. Marom, Adv. Funct. Mater., 2009, 19, 1293 CrossRef CAS.
- B. Adhikari and S. Majumdar, Prog. Polym. Sci., 2004, 29, 699 CrossRef CAS.
- C. Li, H. Bai and G. Shi, Chem. Soc. Rev., 2009, 38, 2397 RSC.
- A. O. Govorov, G. W. Bryant, W. Zhang, T. Skeini, J. Lee, N. A. Kotov, J. M. Slocik and R. R. Naik, Nano Lett., 2006, 6, 984 CrossRef CAS.
- B. C. Sih and M. O. Wolf, Chem. Commun., 2005, 3375 RSC.
- S. K. Bajpai, Y. M. Mohan, M. Bajpai, R. Tankhiwale and V. Thomas, J. Nanosci. Nanotechnol., 2007, 7, 2994 CrossRef CAS.
- D. Muñoz-Rojas, J. Oró-Solé, O. Ayyad and P. Gómez-Romero, Small, 2008, 4, 1301 CrossRef.
- M. M. Oliveira, E. G. Castro, C. D. Canestraro, D. Zanchet, D. Ugarte, L. S. Roman and A. J. G. Zarbin, J. Phys. Chem. B, 2006, 110, 17063 CrossRef CAS.
- A. Postma, Y. Yan, Y. Wang, A. N. Zelikin, E. Tjipto and F. Caruso, Chem. Mater., 2009, 21, 3042 CrossRef CAS.
- J. Ou, J. Wang, J. Zhou, S. Liu, Y. Yu, X. Pang and S. Yang, Prog. Org. Coat., 2010, 68, 244 CrossRef CAS.
- Z. Gao, D. Yap and Y. Zhang, Anal. Sci., 1998, 14, 1059 CrossRef CAS.
- T. A. Morris, A. W. Peterson and M. J. Tarlov, Anal. Chem., 2009, 81, 5413 CrossRef CAS.
- Y. Fu, P. Li, Q. Xie, X. Xu, L. Lei, C. Chen, C. Zou, W. Deng and S. Yao, Adv. Funct. Mater., 2009, 19, 1784 CrossRef CAS.
- R. Makote and M. M. Collinson, Chem. Mater., 1998, 10, 2440 CrossRef CAS.
- B. Yu, D. A. Wang, Q. Ye, F. Zhou and W. Liu, Chem. Commun., 2009, 6789 RSC.
- N. Puvaneswari, J. Muthukrishnan and P. Gunasekaran, Indian J. Exp. Biol., 2006, 44, 618 CAS.
- M. Basu, A. K. Sinha, M. Pradhan, S. Sarkar, Y. Negishi, Govind and T. Pal, Environ. Sci. Technol., 2010, 44, 6313 CrossRef CAS.
- C. Hu, T. Peng, X. Hu, Y. Nie, X. Zhou, J. Qu and H. He, J. Am. Chem. Soc., 2009, 132, 857 CrossRef.
- Y. Wang, D. Li, P. Li, W. Wang, W. Ren, S. Dong and E. Wang, J. Phys. Chem. C, 2007, 111, 16833 CAS.
- H. K. Singh, M. Saquib, M. M. Haque and M. Muneer, Chem. Eng. J., 2008, 136, 77 CrossRef CAS.
- M. M. Alnuaimi, M. A. Rauf and S. S. Ashraf, Dyes Pigm., 2008, 76, 332 CrossRef CAS.
- T. Tatsuma, S.-I. Tachibana, T. Miwa, D. A. Tryk and A. Fujishima, J. Phys. Chem. B, 1999, 103, 8033 CrossRef CAS.
- V. K. Pareek, S. J. Cox, M. P. Brungs, B. Young and A. A. Adesina, Chem. Eng. Sci., 2003, 58, 859 CrossRef CAS.
- S. B. Berman, M. J. Zigmond and T. G. Hastings, J. Neurochem., 1996, 67, 593 CrossRef CAS.
- W. Ye, D. Wang, H. Zhang, F. Zhou and W. Liu, Electrochim. Acta, 2010, 55, 2004 CrossRef CAS.
- X. Qin, H. Wang, Z. Miao, X. Wang, Y. Fang, Q. Chen and X. Shao, Talanta, 2011, 84, 673 CrossRef CAS.
- B. Fei, B. Qian, Z. Yang, R. Wang, W. C. Liu, C. L. Mak and J. H. Xin, Carbon, 2008, 46, 1795 CrossRef CAS.
- C. Guo, P. Boullanger, L. Jiang and T. Liu, Colloids Surf., B, 2008, 62, 146 CrossRef CAS.
- M. D. L. Oliveira, M. T. S. Correia and F. B. Diniz, Synth. Met., 2009, 159, 2162 CrossRef CAS.
- H. Cai, T. M.-H. Lee and I. M. Hsing, Sens. Actuators, B, 2006, 114, 433 CrossRef.
- J. Yu, G. Dai and B. Huang, J. Phys. Chem. C, 2009, 113, 16394 CAS.
- X. Hu, C. Hu, T. Peng, X. Zhou and J. Qu, Environ. Sci. Technol., 2010, 44, 7058 CrossRef CAS.
- L. Kuai, B. Geng, X. Chen, Y. Zhao and Y. Luo, Langmuir, 2010, 26, 18723 CrossRef CAS.
- Y. Li, H. Zhang, Z. Guo, J. Han, X. Zhao, Q. Zhao and S.-J. Kim, Langmuir, 2008, 24, 8351 CrossRef CAS.
- J. J. McMahon, M. Barry, K. J. Breen, A. K. Radziwon, L. D. Brooks and M. R. Blair, J. Phys. Chem. C, 2008, 112, 1158 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1nj20850k |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2012 |