DOI:
10.1039/C1NJ20627C
(Paper)
New J. Chem., 2012,
36, 155-160
Synthesis of metal–carbon core–shell nanoparticles by RAPET (Reaction under Autogenic Pressure at Elevated Temperatures)†
Received
(in Montpellier, France)
18th July 2011
, Accepted 25th October 2011
First published on 17th November 2011
Abstract
We present herein a method to produce metal–carbon core–shell nanoparticles by dissociating the corresponding metal acetates under their autogenic pressure at elevated temperatures. The reaction is a one-stage, solvent-free, catalyst-free, competent, and a very cheap way for the synthesis of metal@carbon nanoparticles with a core–shell structure. The morphology, air-stability, structure, and other properties of the as-prepared Ag@C, Cu@C and Pb@C nanoparticles are demonstrated in this paper. We also provide a criterion for those metals whose acetates would yield metal@carbon structures, as opposed to those metals whose acetates would yield MO@C as the final product.
Introduction
Nanoparticles have found widespread applications in various fields of engineering. The synthesis of nanomaterials is the subject of the current research due to their variety of structures, interesting properties, as well as technological applications in electronics, catalysis, chemical engineering, pharmaceutics, biology, magnetic recording, etc.1,2
Core/shell nanoparticles are nanostructures that have a core made of a material coated with another material. Materials are coated for a number of reasons: coatings can make a substance biocompatible, increase a material's thermal, mechanical or chemical stability, increase its durability, lifetime, decrease friction or inhibit corrosion, avoid aggregation, prevent the spreading of the nanoparticles to the environment, and they may change the overall physicochemical properties of the material. The applications of encapsulated metallic nano- and micro-structures range from catalysis, purification of water wells, food additives,3–5fuel amplifiers,6 electronic devices, as well as the field of energy storage and conversion. One of the main challenges in coating substrates is to achieve a homogeneous layer of the organic/inorganic material without the formation of excess nanoparticles in the nanocomposite. The interest in nanocoatings relies mostly on the combination of the properties of the two (or more) materials involved. An important feature here is that one of the materials (the shell) will determine the surface properties of the particles, while the other (the core) is completely encapsulated by the shell so that it does not contribute to surface properties at all, but can be mainly responsible for other (optical, catalytic, magnetic, etc.) properties of the system. Apart from this extremely important feature, it is also essential to take into account the possible interactions between the core and the shell, which may in certain cases determine the potential applications of the material. Specifically in energy devices, carbon-based materials have been added with the active materials in order to improve the electrical, electrochemical and electrocatalytic properties. Taking into account the unique combination of properties achieved by core–shell geometry, we have demonstrated the important role of in situ-formed amorphous carbon shells in the properties of various metals.
There are various methods for the formation of nanoparticles with core–shell structures; sonochemistry,7,8 electrochemistry,9ammonia catalysis,10 and laser-induced fusion,11 amongst others. Several reports on the formation of core–shell nanostructures of transition metals have already been published.12–16 For example, Tang et al.14 used a MW-assisted method17 to prepare Ag–C core–shell particles. The size distribution of particles obtained by this method is wide, namely, ranging from 200 to 800 nm. The typical diameter is 660 nm. In the work of L. S. Wang et al.15 Ag-core/C-shell nanocables were synthesized via a hydrogen arc method.15,18 This structure was formed under H2 pressure, a current of 100 A, and a voltage of 25 V. The method of L.-B. Luo et al.16 allows the achievement of a silver@carbon-rich composite by a preparation method that took several days. The RAPET method is a novel, one-step, solvent-free and very simple technique for the fabrication of spherical core–shell nanoparticles.17,18 The chemical dissociation and transformation reaction takes place under the autogenic pressure of the precursor at a certain temperature, followed by the gradual cooling of the reactor to room temperature. Herein we report on obtaining Ag@C, Cu@C, and Pb@C from the corresponding acetates using the RAPET method. The current results are compared with previous metal@carbon products resulting from the decomposition of various metal–organic compounds yielding also metal cores coated by carbon.17–22 The RAPET was conducted at high temperatures (700–1000 °C); however, the decomposition mechanism involves several stages. The decomposition of silver, copper and lead acetates occurs at 150–250 °C.17,18,21 All three acetates decompose into their corresponding oxides, which are further reduced by carbon monoxide to the metallic state, as proposed elsewhere.23–26Ag@C and Cu@C were found to remain stable over a long time, whereas Pb tends to oxidize after one day under ambient conditions. Unlike the acetates of the noble metals, the acetates of the stronger metals (e.g., Zn and Mg) yield a metal oxide/carbon core/shell structure. We therefore propose a “RAPET Rule” that relates the standard reduction potential of the metal in the acetate salt to the nature of the final product (metal or metal oxide).
Experimental setup
Synthesis
All the precursors, silver acetate (99% purity), copper(II) acetate (98% purity), and lead (IV) acetate (95% purity) were purchased from Sigma Aldrich and used as received, without further purification. Each material was inserted into a cell assembled from stainless steel Swagelok parts. A “1/2” union part was capped from both sides by standard plugs (Fig. 1). The volume of the cell was 4.3 ml. For these reactions, 0.3 g (1.80 × 10−3 mol) of silver acetate, 3.41 g (18.77 × 10−3 mol) of copper acetate and 4.3 g (9.70 × 10−3 mol) of lead acetate were inserted separately into the cell at room temperature under nitrogen in a glove box. In addition, experiments were carried out with a large amount of precursors (1.5 g of silver acetate, 7.8 g of copper acetate and 8.5 g of lead acetate) for checking the influence of material quantity on the morphology. The results are shown below.
 |
| | Fig. 1 An overview of Swagelok used for the RAPET reaction. | |
The filled cell was closed and placed inside an iron pipe in the middle of the furnace. The temperature of the furnace was raised to 850 °C at a rate of 10 °C min−1. The cell was kept at 850 °C for three hours. At the end of the reaction, the Swagelok was cooled to room temperature, opened, and the black powders of Ag@C (0.205 g, 68.1% yield), Cu@C (1.173 g, 34.4% yield) and Pb@C (1.989 g, 45.6% yield) were obtained. The yields of the obtained materials are very close to the weight percentage of metal atoms in the metal-acetate salts (64.7% Ag in silver acetate, 34.9% Cu in copper(II) acetate and 46.7% Pb in lead(IV) acetate). This indicates that the main component remaining at the end of the RAPET reaction is the metallic core, and most of the C, H, and O in the acetates are released as hydrocarbon gases, CO, and CO2. This result is further substantiated by the C, H, N, O and TGA analysis shown below. The weight percentage of carbon in all obtained M@C materials is less than 13%, and therefore the good correlation between the yields and the weight percentage of metals in the M@C structures is very reasonable. The morphological and crystal structure characteristics, elemental content, stability at high temperature and other properties of the above materials are presented in the Results and Discussion section.
Characterization
To probe the nature of the product and the purity of its crystal structure, the X-ray diffraction patterns were measured with a Bruker AXS D* Advance powder X-Ray diffractometer, Cu–Kα radiation, wavelength: 1.5406 Å was used. To determine the content of the C, H, and O in all the products, elemental analysis was carried out employing an Eager 200 C, H, N analyzer and an EA1110 Oxygen analyzer. The morphologies and nanostructures of the as-prepared products were characterized with a Scanning Electron Microscope (SEM) Model JSM-840, a Transmission Electron Microscope (TEM) Model JEM-1200EX and a High Resolution TEM Model JEOL-2010, working at acceleration voltages of 80 (TEM) and 200 KeV (HRTEM). Samples for TEM and HRTEM were prepared by ultrasonically dispersing the products in isopropanol, by placing a drop of this suspension onto a copper grid (for Ag and Pb) and a gold grid (for Cu) coated with an amorphous carbon film, and then drying under air. The study of thermal decomposition of these three precursors (Ag, Pb and Cu acetates) and products was performed with a TGA Q500 instrument (TA Instruments, USA) with the following parameters: heating rate 10 °C min−1, nitrogen flow 120 ml min−1, sample mass about 4–10 mg. Raman spectroscopy was used to define the nature of the carbon that forms the shell in the metal@carbon composite for all three metals. An Olympus BX41 (Jobin Yvon Horiba) Raman spectrometer was employed, using the 514.5-nm line of an Ar laser as the excitation source. The impedance of the core–shell Ag@C material was measured at a frequency range of 50 kHz–500 Hz using a frequency response analyzer model-1255 from Solartron, Inc. driven by ZPlot software (Scribner Associates, Inc.). The results obtained at 30° and 60 °C are presented in the form of Z′, (-Z′′)-plots (Z′ and -Z′′ are real and imaginary parts of the complex impedance). For these measurements, a pellet made from the Ag@C material (∼1 cm in diameter, the density of pellet is 4.75 gr cm−3, the pressure applied for pelletizing was 5 tsi) was placed between two polished stainless steel discs and mounted in the parts of the 2325 coin-type cell (from NRC, Canada). For comparison, similar impedance measurements were performed with silver nano-sized particles of around 50 nm, at 30 °C. These silver nanoparticles were synthesized in our lab. All the measurements were carried out in a thermostat. Fig. 1S (in ESI†) shows the SEM micrographs of the pellet. The broken pellet was also analyzed by SEM, and the picture was taken at 90 degrees. Particles' size ranges from tens of nanometres to few microns.
Results and discussion
In this section we present results related to the different metal acetates that produce particles with a core–shell structure of the metal coated with carbon.
Morphology (SEM and HR-TEM analysis)
The transmission electron micrographs at high resolution of all the products are shown in Fig. 2.
 |
| | Fig. 2
Transmission electron micrographs of (a) Ag@C, (b) Cu@C and (c) Pb@C core–shell nanoparticles at high resolution. | |
Fig. 2 presents a part of spherical metallic particles (dark areas) of Ag, Cu and Pb coated by the carbon shell (brighter areas). Moreover, Fig. 2a and b depict the perfect arrangement of atomic layers of Ag and Cu that can also prove their crystallinity that is covered by an amorphous or graphitic carbon layer. Fig. 2c shows Pb@C nanostructures with a lower magnification than that of Ag@C and Cu@C because the electron beam of TEM can melt Pb particles due to their low melting point (327 °C). The diameter of the Ag@C (a) particles is in the 50–150 nm range (the thickness of the carbon shell is in the range of 5–15 nm), while the range for the Cu@C (b) particles is 50–200 nm (the thickness of the shells is in the range of 10–20 nm). The size of the Pb@C (c) particles is 50–400 nm (the shells are in the range of 0–50 nm). Fig. 2S(a)–(c) (ESI†) show aggregates that are obtained in experiments with a large quantity of the precursor. In this case the autogenic pressure that was created in the cell is higher and aggregated particles were obtained.
Fig. 3 shows the SEM image of the obtained particles and illustrates that most of the products have a spherical shape. Their sizes vary between 10 nm and 500 nm, similar to the results obtained from TEM measurements. The picture shows the mode of aggregation. While the spherical contour of the particles is well observed for the silver and lead agglomerates, flat surfaces, and even hexagonal shapes are observed for the Cu@C products. A transparent shell surrounds the lead cores in Fig. 3c, emphasizing the core–shell nature of the product.
 |
| | Fig. 3
Scanning electron micrographs of (a) Ag@C, (b) Cu@C and (c) Pb@C core–shell nanoparticles. | |
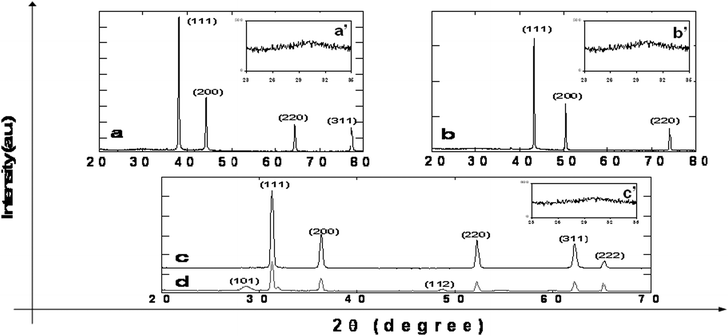
![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m). The peak positions match the PDF file for Ag (PDF: 01-089-3722). The XRD pattern of the as-prepared Cu@C core–shell particles is presented in Fig. 4b. The diffraction peaks observed at 2θ = 43.32°, 50.45° and 74.13° are assigned as (111), (200) and (220) crystal planes of the face-centered cubic phase of Cu (space group: Fmm). The peak position matches the PDF file for Cu (PDF: 01-085-1326). The XRD pattern of the as-prepared Pb@C core–shell particles is presented in Fig. 4c. The diffraction peaks observed at 2θ = 31.28°, 36.27°, 52.23°, 62.16° and 65.25° are assigned as (111), (200), (220), (311) and (222) crystal planes of the face-centered cubic phase of Pb (space group: Fmm). The peak position matches the PDF file for Pb (PDF: 03-065-2873). All the products were checked again by XRD after aging under an ambient atmosphere for a certain time period (from one day to a week). Some lead particles were oxidized, and Fig. 4d shows the reflections of both lead and lead oxide. The diffraction peaks of lead oxide observed at 2θ = 28.63°, 31.83°, 35.74°, 48.60° and 54.76° are assigned as (101), (110), (002), (112) and (211) crystal planes of the tetragonal phase of PbO (space group: P
m). The peak positions match the PDF file for Ag (PDF: 01-089-3722). The XRD pattern of the as-prepared Cu@C core–shell particles is presented in Fig. 4b. The diffraction peaks observed at 2θ = 43.32°, 50.45° and 74.13° are assigned as (111), (200) and (220) crystal planes of the face-centered cubic phase of Cu (space group: Fmm). The peak position matches the PDF file for Cu (PDF: 01-085-1326). The XRD pattern of the as-prepared Pb@C core–shell particles is presented in Fig. 4c. The diffraction peaks observed at 2θ = 31.28°, 36.27°, 52.23°, 62.16° and 65.25° are assigned as (111), (200), (220), (311) and (222) crystal planes of the face-centered cubic phase of Pb (space group: Fmm). The peak position matches the PDF file for Pb (PDF: 03-065-2873). All the products were checked again by XRD after aging under an ambient atmosphere for a certain time period (from one day to a week). Some lead particles were oxidized, and Fig. 4d shows the reflections of both lead and lead oxide. The diffraction peaks of lead oxide observed at 2θ = 28.63°, 31.83°, 35.74°, 48.60° and 54.76° are assigned as (101), (110), (002), (112) and (211) crystal planes of the tetragonal phase of PbO (space group: P![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) nmm). These values are in good agreement with the diffraction peaks, peak intensities and cell parameters of crystalline PbO (PDF: 00-005-0561). Relative to the crystallinity of the metals the carbon material is amorphous as revealed in the XRD patterns. But when the y-axis is magnified and viewed, in the range of 0–500 a.u. we can see a broad peak in the 2θ range of 24–32° which is typical of amorphous carbon. This peak is detected as shown in Fig. 4a′–c′4.
nmm). These values are in good agreement with the diffraction peaks, peak intensities and cell parameters of crystalline PbO (PDF: 00-005-0561). Relative to the crystallinity of the metals the carbon material is amorphous as revealed in the XRD patterns. But when the y-axis is magnified and viewed, in the range of 0–500 a.u. we can see a broad peak in the 2θ range of 24–32° which is typical of amorphous carbon. This peak is detected as shown in Fig. 4a′–c′4.
 |
| | Fig. 4
XRD patterns of (a) Ag, (b) Cu, (c) Pb nanoparticles measured immediately after annealing at 850 °C for 3 hours, and (d) Pb sample presented in 4c left to age for one day under ambient conditions. a′, b′ and c′ represent the presence of amorphous carbon. | |
C, H, N, O and TGA analysis
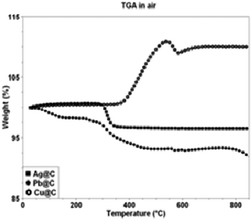
The carbon, hydrogen, nitrogen and oxygen contents in the products are determined by elemental analysis measurements (Table 1). The weight percentages of carbon in Ag@C, Pb@C and Cu@C core–shell nanoparticles are 4.16, 7.48 and 12.63, of hydrogen 0.02, 0.09 and 0.15, and of oxygen 1.74, 4.93 and 0.30, respectively. The results of the TGA measurements conducted under air of all the products allow us to probe their stability at high temperatures.
Table 1 The weight percentages of metal, carbon, hydrogen and oxygen in Ag@C, Pb@C and Cu@C samples obtained from the elemental analysis
| Sample name |
%C |
%H |
%O |
%Metal |
|
Ag@C
|
4.16 |
0.02 |
1.74 |
94.08 |
|
Pb@C
|
7.48 |
0.09 |
4.93 |
87.50 |
|
Cu@C
|
12.63 |
0.15 |
0.30 |
86.92 |
The results are shown in Fig. 5.
A weight loss of ∼4% was detected for the Ag@C core–shell structure at a temperature range of 25–350 °C. This percentage is approximately the amount of carbon in the product, and the weight loss is due to carbon combustion. The constant mass of the Ag@C at 350 °C to 800 °C indicates that perhaps a thin layer of silver oxide is preventing further oxidation which would have increased the weight. This layer also prevents the combustion of the carbon, which would have resulted in a weight loss. The remaining metal weight at 800 °C fits well the number in Table 1. TGA analysis of Cu@C particles shows a weight increase of 10% in air. The maximum weight increase is reached at 500 °C and remains constant at higher temperatures. The increase in weight is apparently due to the formation of CuO. If all the copper would have been oxidized, an increase of about 9% is expected, which is close to the detected weight gain. Combustion of carbon leading to the formation of CO2 occurs as well, as indicated by the TGA measurement coupled to mass-spectrometry (see picture 3S in ESI†). Pb@C was also measured by TGA a few days after the material was prepared and stored under ambient environment. During this storage period, the composite was oxidized, and therefore the examined sample by TGA analysis was PbO@C rather than Pb@C. PbO@C particles showed a weight decrease at 25–400 °C due to carbon combustion. The weight percentage of the material at 400–800 °C was ∼92% of the starting weight of the PbO@C particles and is in good agreement with total PbO percentages at C, H, N and O measurements (∼92%, see Table 1).
Raman measurement
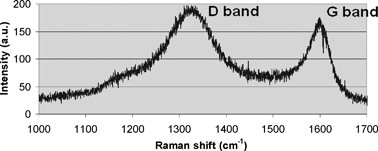
Fig. 6 presents the Raman spectrum and shows two peaks related to carbon at 1332 cm−1 and 1597 cm−1 (D and G bands, respectively). The G band is related to graphitic carbon and the D band to disordered carbon.
Impedance measurements
In Fig. 7a, we present Z′, (-Z′′)-plots obtained from the Ag@C material at 30° and 60 °C. The results of three measurements at each temperature during several hours demonstrate a sufficient reproducibility. We can conclude that the impedance of the material is slightly dependent on the frequency. As expected, the impedance of the core–shell Ag@C material at the elevated temperature (60 °C) is lower than at 30 °C (∼0.18 Ohm vs. ∼0.28 Ohm, respectively, in the frequency range of 2.5 kHz–500 Hz). Fig. 7b is a Z′, (-Z′′)-plot measured for silver nanoparticles at 30 °C (from the as-prepared pellet) after 3 and 72 hours. The impedance of the Ag nanoparticles does not depend upon the frequency, but increases substantially with aging time, indicating some possible reactions of the nanoparticles and the formation of the surface species due to adsorbed water and oxygen.27 From the results obtained we conclude that the Ag@C core–shell material synthesized in this work possesses low impedance comparable with that of the nano-Ag particles (around 0.2–0.23 Ohm, Fig. 7b). We also suggest that the stable impedance of the Ag@C particles at both temperatures is related, to some extent, to the protective properties of the carbon shell that may prevent reactions of the metal core in air.
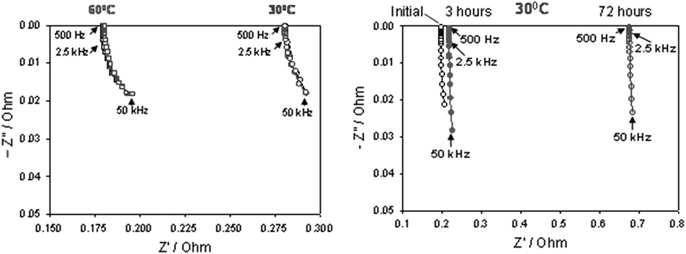 |
| | Fig. 7 (a) Resistance plots of the core–shell Ag@C material measured at 30 °C and 60 °C. (b) Resistance plots of the nano-Ag particles measured at 30 °C initially and after 3 and 72 hours. | |
RAPET rule
All the transition metal acetates in the current study underwent thermal decomposition in the RAPET system. As a result, carbon-coated Ag, Cu and Pb nanoparticles are obtained. For all three acetates their thermal decomposition takes place at temperatures of 150–300 °C.17,18,21 As explained above, the metal oxides are intermediates that are further reduced by CO to form the zero valent metals. Later on the metals form the core because of their higher solidification rate than carbon, and carbon forms the shell layer.17 This mechanism was published in previous studies17,27,28 based on TGA and MS analysis and is as follows:| | | M(CH3COO)2 → MCO3 + CH3COCH3 | (1) |
where M = Ag, Cu or Pb. Stoichiometric relationships vary according to the metal. Reactions (1)–(3) occur at 250–350 °C. Reaction (4′) takes place at all the temperature ranges but becomes more kinetically and thermodynamically favorable with an increase in temperature. The value of the Gibbs free energy of reaction (4′′) is negative at temperatures below 700 °C. Therefore, at temperatures higher than 700 °C, the only occurring reaction is 4′. When the reaction system is cooled (in the end of the reaction), the carbon shell is formed around the metallic particles (core) at temperatures below 700 °C. The cooling reduced the solubility of the carbon in the metal and the carbon phase is separated from the metal forming a ring around the core. In previous17 and current works of our research group, the following results were recorded: metals with a reduction potential lower than −0.4 V generated their corresponding oxides as core, and metals with a reduction potential higher than −0.4 V reached their metallic form. The values of free energy ΔG0 for each metal are calculated to form the respective metals from the corresponding oxide in the presence of carbon monoxide. The results are shown in Table 2.
Table 2
Reduction potentials and calculated thermodynamic data for all the metals
| Element |
Co
|
Ni
|
Fe
|
Ag
|
Cu
|
Pb
|
Zu
|
Bi
|
|
Reduction potential/V |
−0.28 |
−0.25 |
−0.44 |
0.8 |
0.34 |
1.57 |
−0.79 |
0.32 |
| Gibbs free energy of reaction (4′)/kJ mol−1 |
−41 |
−49 |
14 |
−238 |
−130 |
−73 |
55 |
−282 |
| Result |
Metal |
Metal |
Oxide
|
Metal |
Metal |
Metal |
Oxide
|
Metal |
The positive values of free energy for Fe and Zn support our assumption that thermodynamically, the reduction to the corresponding metallic state is not favorable for these metals; therefore the results of the RAPET reactions for these metals (Fe and Zn) are the corresponding oxides. We called this regularity the “RAPET Rule”—stating that the acetates of strong metals like the alkali, alkaline earth, rare earth metals with a reduction potential lower than −0.4 V would form a core–shell structure composed of a metal oxide core. The acetates of the noble metals, as well as metals with a standard reduction potential larger than −0.4 V, will yield a product having a metal as a core.
Conclusions
RAPET is a method developed in our research group. It has many outstanding advantages; this non-aqueous, template-, surfactant-, catalyst- and solvent-free method allows us to synthesize metal–carbon core–shell nanoparticles. In the study of the RAPET of metal acetates, we have demonstrated a direct relation between the reduction potential of the metal and the product obtained from the reaction. The “RAPET rule” enables us to predict accurately which product will be obtained—metal or metal oxide coated by carbon.
Notes and references
- N. Sounderya and Y. Zhang, Recent Pat. Biomed. Eng., 2008, 1, 34–42 CrossRef CAS.
-
R. C. Merkle, “Nanotechnology is coming”, Frankfurter Allgemeine Zeitung, 2000, 55.
- E. Pena, A. Sanchez, Z. Portillo and L. Solano, Arch. Latinoam. Nutr., 2003, 53, 133–140 CAS.
- D. Ragland, J. L. Schneider, S. F. Amass and M. A. Hill, J. Swine Health Prod., 2006, 14, 82–88 Search PubMed.
- M. Saric, N. Filipovic, N. Hladni and O. Grujic, Acta Aliment., 1998, 27, 21–27 Search PubMed.
- Y. V. Frolov, P. Pokhil and V. S. Logachev, Combust., Explos. Shock Waves, 1972, 8, 168–187 CrossRef.
- V. G. Pol, H. Grisaru and A. Gedanken, Langmuir, 2005, 21, 3635–3640 CrossRef CAS.
- Y. Koltypin, N. Perkas and A. Gedanken, J. Mater. Chem., 2004, 14, 2975–2977 RSC.
- X. Li,
et al.
, Chem. J. Chin. Univ., 2005, 26, 1869–1872 CAS.
- P. Wang, D. Chen and F. Tang, Langmuir, 2006, 22, 4832–4835 CrossRef CAS.
- V. Prashant, J. Phys. Chem. B, 2002, 106, 7729–7744 CrossRef.
- S. V. Pol, V. G. Pol and A. Gedanken, Chem.–Eur. J., 2004, 10, 4467 CrossRef CAS.
- V. G. Pol, G. Seisenbaeva, V. G. Kessler and A. Gedanken, Chem. Mater., 2004, 16, 1793 CrossRef.
- S. Tang, S. Vongehr, Z. Zheng, H. Liu and X. Meng, J. Phys. Chem. C, 2010, 114, 18338–18346 CAS.
- L. S. Wang, D. B. Buchholz, Y. Li, J. Li, C. Y. Lee, H. T. Chiu and R. P. H. Chang, Appl. Phys. A: Mater. Sci. Process., 2007, 87, 1–6 CrossRef CAS.
- L.-B. Luo, S.-H. Yu, H.-S. Qian and J.-Y. Gong, Chem. Commun., 2006, 793–795 RSC.
- S. C. Tang, S. Vongehr and X. K. Meng, J. Phys. Chem. C, 2010, 114, 977 CAS.
- X. K. Wang, X. W. Lin, M. Mesleh, M. F. Jarrold, V. P. Dravid, J. B. Ketterson and R. P. H. Chang, J. Mater. Res., 1995, 10, 1 CrossRef CAS.
- S. V. Pol, V. G. Pol and A. Gedanken, Chem.–Eur. J., 2004, 10, 1–8 CrossRef.
-
V. G. Pol, S. V. Pol and A. Gedanken, Properties of Nanostructured Metal Oxides Synthesized by the RAPET Approach, Metal Oxide Nanostructures and Their Applications, American Scientific Publishers, 2008 Search PubMed.
- A. Gedanken and E. Luvchik, Eur. J. Inorg. Chem., 2008, 15, 2471–2475 CrossRef.
- S. V. Pol, V. G. Pol and A. Gedanken, J. Phys. Chem. C, 2007, 111, 13309–13314 CAS.
- V. Logvinenko, O. Polunina, Yu. Mikhailov, K. Mikhailov and B. Bokhonov, J. Therm. Anal. Calorim., 2007, 90(3), 813–816 CrossRef CAS.
- A. Y. Obaid, A. O. Alyoubi, A. A. Samarkandy, S. A. Al-Thabaiti, S. S. Al-Juaid, A. A. El-Bellihi and El-H. M. Deifallah, J. Therm. Anal. Calorim., 2000, 61, 985–994 CrossRef CAS.
- M. A. Mohamed, S. A. Halawy and M. M. Ebrahim, Thermochim. Acta, 1994, 236, 249–262 CrossRef CAS.
- S. Pol, V. Pol, I. Felner and A. Gedanken, Eur. J. Inorg. Chem., 2007, 14, 2089–2096 CrossRef.
- R. Janardhanan, M. Karuppaiah, N. Hebalkar and T. N. Rao, Elsevier, 2009, 28, 2522–2530 CAS.
- E. Holodelshikov, I. Perelshtein and A. Gedanken, Inorg. Chem., 2011, 50, 1288–1294 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1nj20627c |
|
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2012 |
Click here to see how this site uses Cookies. View our privacy policy here.