DOI:
10.1039/C1NJ20252A
(Paper)
New J. Chem., 2012,
36, 139-147
Synthesis of mesoporous chromium phosphatesvia solid-state reaction at low temperature
Received
(in Montpellier, France)
18th March 2011
, Accepted 22nd September 2011
First published on 15th November 2011
Abstract
Using solid-state reaction at low temperature (SSRLT), mesoporous chromium phosphates were successfully synthesized in the presence of a surfactant template, cetyltrimethyl ammonium bromide (CTAB), at a temperature of 353 K without using a water solvent. The synthesized chromium phosphates were characterized by XRD, EDS, HR-TEM, N2-physisorption, TGA-DSC, FT-IR, and UV-vis spectroscopies. It was found that mesoporous chromium phosphate can be formed only at atomic ratios of P/Cr ≥ 1.8. The mesophase at P/Cr = 2.0 showed the highest ordering of pore arrays, with a specific surface area as high as 250.78 m2 g-1 and an average pore size of 3.48 nm. The catalytic performance of the synthesized chromium phosphates was examined using the dehydration of isopropanol to propene as the model reaction. The results indicated that the mesoporous chromium phosphates synthesized via the SSRLT route exhibited significantly higher isopropanol conversions and propene yields than that synthesized via the solution sol–gel route and the non-mesoporous chromium phosphate.
1. Introduction
Chromium
phosphates are of considerable importance, owing to their versatile applications in the areas of sorption/ion exchange of divalent metal ions,1,2 and catalysis of various organic reactions, such as, the dehydration of alcohols,3–5 the oxidative dehydrogenation of alkanes,4,6,7 the isomerization of alkenes,7,8 and the alkylation of aromatics.3–9 The structure and composition of the chromium phosphates have been found to be dependent on the synthesis route. A series of chromium phosphates, such as amorphous CrPO4,2,3,10 and crystalline Na2CrP2O7·0.5H2O,11 Cr(H2O)4HP2O7·3H2O,12 and CrPO4·6H2O,13 have been synthesized via wet chemistry routes, such as the sol-gel method and the hydrothermal synthesis. There are also a few reports on the synthesis of chromium phosphatesvia the solid state reaction at high-temperature (usually higher than 500 °C), which generated the crystallines α-CrPO4,14, 15β-CrPO4,10,16Cr(PO3)3,17Cr3(PO4),18Cr7(PO4)6,19Cr6(P2O7)4,21 and Na17Cr9P12O58H12.22 All these compounds are usually non-porous or possess low specific surface areas and pore volumes and are thus unsuitable for catalytic and ion exchange applications. Because of their unique properties, such as high specific surface area and pore volume, tunable nanopore size, adjustable and versatile framework composition, and controllable particle morphology, the mesoporous inorganic materials have attracted much interest. In the synthesis of mesoporous materials, the solution sol-gel technique is usually employed. The controlled hydrolysis of inorganic precursor species and their subsequent condensation, templated by the supramolecular assembly of surfactant micelles, constitute the key steps. Through this route, mesoporous silicas, including M41S,23SBA-n,24MSU-X,25 and HMS,26 and various single-component non-silica mesoporous materials of metal oxides,27-29 were successfully synthesized. However, the application of solution sol-gel method in the synthesis of multi-component non-silica mesoporous materials was not successful so readily, although a few mesoporous mixed metal oxides,30-32 and aluminophosphates,33 have been reported in the literature. The main difficulties may come from the control of hydrolysis of the precursor species to match their condensation, and from simultaneous organization of different units of the framework components on the surface of surfactant assemblies, in order to generate a stable mesoporous phase with all framework components in the desired proportion and a homogeneous distribution at molecular level. Accordingly, a complex and elaborate procedure has to be employed for the synthesis of multi-component non-silica mesoporous materials. For example, Tarafdar et al34 synthesized the mesoporous chromium (III) phosphate using an unconventional sol-gel technique, comprising of the reduction of ammonium dichromate with ethanol and nitric acid, the formation of gel in the presence of ammonium dihydrogen phosphate, urea and tetradecyltrimethylammonium bromide (TTBr) as structure directing agent and the programmed hydrothermal treatment at 353 K for 3 days, 363 K for 2 days and 383 K for 1 day. The obtained chromium phosphate possessed a specific surface area of 201 m2g−1. In addition, the solution sol-gel method provides a relatively low yield of mesoporous materials and produces a large amount of waste water. In 2004, Lu et al35 reported the synthesis of MSU-mesostructured ZrO2via a solid-state reaction route, which involves the milling of zirconyl chloride together with nonionic block-copolymer surfactant and then their reaction with sodium hydroxide under vigorous stirring, followed by heating at ca. 423 K. This work is of interest, as it may present an alternative approach to the synthesis of mesoporous materials. Since then, however, no other mesoporous material has been reported to be synthesized via the solid-state reaction. We report in this paper that mesoporous chromium phosphates, possessing better textural properties than those via the solution sol-gel route in ref. 34, can be synthesized via the route of solid-state reaction at low temperature (SSRLT) and under atmospheric pressure. It is of note that the SSRLT offers a few advantages. Firstly, the temperature employed in the SSRLT route is as low as that for the solution sol-gel route, which is significantly lower than that for the conventional solid-state reaction in the synthesis of non-porous inorganic material such as chromium phosphates. Secondly, compared to the solution sol-gel route, the SSRLT route involves fewer steps and produces less waste water but a higher yield of mesoporous materials. Thirdly, when employed as the catalyst for the dehydration of isopropanol to propene, the mesoporous chromium phosphate synthesized via the SSRLT route is significantly more active and selective than that prepared via the solution sol-gel route or the non-mesoporous chromium phosphate prepared with the conventional sol-gel route.
2. Experimental
2.1. Synthesis
Synthesis of mesoporous chromium phosphatesvia the SSRLT route.
Various P/Cr molar ratios of CrCl3·6H2O (A. R., Sigma Aldrich) and NaH2PO4·2H2O (A. R., Sigma Aldrich) were mixed together and ground in a mortar for 20 min and then mixed further with a selected amount of cetyltrimethyl ammonium bromide (CTAB, A. R. Sigma Aldrich) and ground together for another 15 min. The obtained mixtures with atomic ratios of P![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Cr:CTAB = x:1.0:0.001, where x = 1.0, 1.6, 1.8, 2.0, 2.2, 2.4, and 3.0, were transferred into beakers for heating at 373 K for 24 h. After cooling to room temperature, the samples were washed thoroughly with distilled water and then dried at 373 K for 24 h. To remove the surfactant, the samples were further calcined in N2 at 823 K for 6 h.
:Cr:CTAB = x:1.0:0.001, where x = 1.0, 1.6, 1.8, 2.0, 2.2, 2.4, and 3.0, were transferred into beakers for heating at 373 K for 24 h. After cooling to room temperature, the samples were washed thoroughly with distilled water and then dried at 373 K for 24 h. To remove the surfactant, the samples were further calcined in N2 at 823 K for 6 h.
Synthesis of non-mesoporous chromium phosphatesvia the conventional sol–gel route4,10.
A solution containing chromium and phosphate was first prepared by dissolving CrCl3·6H2O and NaH2PO4·2H2O with atomic ratio of P:Cr = 2.0 into distilled water at 273 K, and then a cold ammonium aqueous solution (25% volume) was added with continuous stirring. The resulting precipitation was aged at room temperature for 24 h, then washed thoroughly with distilled water, and dried at 373 K for 24 h. Finally, the synthesized chromium phosphates were calcined at 823 K for 6 h.
Synthesis of mesoporous chromium phosphatesvia the solution sol–gel route34.
A solution containing 2.52 g of ammonium dichromate and 25 ml water was first mixed with 8 ml ethanol and 8 ml HNO3 under stirring, and then another solution which contained 4.60 g ammonium dihydrogen phosphate, 1.46 g cetyltrimethyl ammonium bromide (A. R. Sigma Aldrich) and 29 ml water was added with continuous stirring. After that, 10 g of urea was added to the mixture. The resulting mixture, having an atomic ratio of P : Cr : CTAB = 2.0 : 1.0 : 0.4, was aged at 353 K for three days and then transferred into a teflon-lined autoclave to be subjected to hydrothermal treatment at 363 K for two days and 373 K for one day. The solid was recovered by filtration and dried at 333 K. Finally, the synthesized sample was thoroughly washed with ethanol and calcined at 823 K for 6 h.
2.2. Characterization
The structure of the synthesized compounds was determined using X-ray diffraction (XRD) (Brucker D8 advance diffractometer, Cu-Kα radiation, at 1.5406 Å). The diffraction data were recorded with a scanning speed of 0.02° s−1 for 2θ angles between 0.35° and 75°.
The morphology of the synthesized compounds was examined using high resolution transmission electron microscopy (HR-TEM) (JOEM-3010 electron microscope). The local composition of the synthesized compounds was analyzed by energy dispersible X-ray spectroscopy (EDS) with an elemental analysis accessory (OXFORD).
The BET surface area and porosimetry of the synthesized compounds were measured using nitrogen adsorption–desorption (Beckman Counter SA 3100). The pore size distribution was calculated using the BJH method from the adsorption branch data.36
The formation of CTAB micelles was examined using Fourier transform infrared (FT-IR) spectroscopy (Varian 3100 spectrometer with a DTGS detector and a CeI beamsplitter). Before the measurement, the sample was ground together with KBr and pressed into a wafer. The data, ranging from 300–4000 cm−1, were recorded as the accumulated result of 20 scans at a resolution of 4 cm−1.
The oxidation state of chromium in the chromium phosphates was determined using ultraviolet-visible diffuse reflection (UV-DR) spectroscopy (Perkin Elmer Lambda 35 UV-vis spectrometer). The data were collected between 200–800 nm using BaSO4 as a reference.
The thermal stability of the synthesized compounds was characterized using thermo-gravimetry analysis (TGA) and differential scanning calorimetry (DSC) (Netzsch STA449C). The sample was heated from room temperature to 1273 K at a rate of 5 K min−1, with a 98.5% N2carrier gas at a flux of 50 ml min−1.
2.3.
Catalytic performance evaluation
To evaluate the catalytic performance of the synthesized chromium phosphate compounds, the dehydration of isopropanol to propene was used as a model reaction. The reaction was performed in a fixed-bed flow-type quartz reactor under the following conditions: catalyst amount = 1.0 g, liquid flow rate of isopropanol (≥99.7%) = 0.085 ml min−1, reaction temperature = 473–563 K (at 30 K intervals), and atmospheric pressure. The effluent from the reactor was cooled in an ice bath, in which the gaseous products and residual isopropanol were separated. After the reaction proceeded smoothly for 60 min, both the liquid and gaseous products were sampled and analyzed. The identification of the products was achieved using a Varian Saturn-2200 GC-MS equipped with a DB-1 capillary column (30 m × 0.25 mm × 0.25 μm). The quantitative analysis of the products was performed using a Perkin Elmer Clarus 500 GC equipped with a thermal conductivity detector (TCD) and a HayeSep DB column (30′ × 1/8′′ × 0.085′′). The conversion of isopropanol and the selectivities to the reaction products were calculated as shown in the Appendix.
3. Results and discussion
3.1 Synthesis of chromium phosphates
For the chromium phosphates synthesized via the conventional sol-gel route and via the solution sol-gel route, XRD characterization (data not shown here) indicates that these two samples are non-mesoporous and mesoporous, respectively, being consistent with refs.10 and 34. Fig. 1 shows the low angle XRD patterns of the chromium phosphates synthesized via the SSRLT route from the initial batches with various P/Cr atomic ratios, before the removal of surfactant. For the samples with an atomic ratio P/Cr < 1.8, no XRD diffraction peak (not shown in the figure) was detected, indicating the absence of mesophase. However, for the samples with an atomic ratio P/Cr ≥ 1.8, a distorted mesophase is identified, as evidenced by a single diffraction peak in the XRD patterns (Fig. 1a–e). With increasing P/Cr molar ratio, the width of diffraction peak increases but its 2θ angle decreases, indicating the broadening of mesopore size distribution and the increase of d-spacing (i.e., pore-to-pore distance; see Table 1). This may be due to the enlargement of mesopores and/or the thickening of the walls of mesopores. The intensity of diffraction peak increases with increasing P/Cr atomic ratio, attaining a maximum at the atomic ratio P/Cr = 2.0, followed by a decrease. After the surfactant removal, the mesostructure is retained in all the samples (see the XRD patterns in the inset in Fig. 1), and still the sample with the atomic ratio P/Cr = 2.0 possesses the high intensity of diffraction peak. Apparently, the sample with the atomic ratio P/Cr = 2.0 possesses the maximum ordering of the mesopore array. The d-spacing of the samples after the surfactant removal (seen in Table 1) decreases from 4.04–5.17 nm to 4.03–4.29 nm, as compared with that before the surfactant removal. This probably occurs because the surfactant removal during the high temperature calcination is accompanied by the condensation of the chromium and phosphate species or their dehydration, resulting in the partial collapse of mesophase and shrinkage of mesopores. The wide angle XRD was also carried out for all the samples, but no diffraction peak was detected (data not shown here), suggesting that all the samples are amorphous.
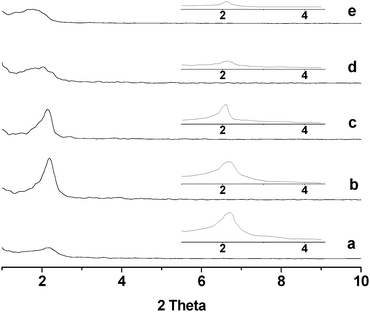 |
| | Fig. 1
XRD patterns of chromium phosphate samples synthesized from initial batches with P/Cr atomic ratios of (a) 1.8, (b) 2.0, (c) 2.2, (d) 2.4, and (e) 3.0 before the surfactant removal and after the surfactant removal (inset). | |
Table 1 The d-spacings in Å obtained from XRD spectra and the O/P and P/Cr molar ratios obtained from EDS for chromium phosphate samples synthesized from initial batches with various P/Cr atomic ratios
| |
P/Cr in the initial batches |
| 1.00 |
1.60 |
1.80 |
2.00 |
2.20 |
2.40 |
3.00 |
|
before surfactant removal;
after surfactant removal;
N.D.: not determined;
N.A.: not available.
|
| P/Cr in the samples |
1.06 |
N.D.c |
1.79 |
2.00 |
N.D. |
2.31 |
3.03 |
| O/P in the samples |
3.66 |
N.D. |
4.76 |
4.69 |
N.D. |
4.71 |
3.89 |
| d-spacing of the samplesa |
N.A
|
N.A.d |
4.04 |
4.13 |
4.17 |
4.52 |
5.17 |
| d-spacing of the samplesb |
N.A
|
N.A
|
4.03 |
4.07 |
4.14 |
4.26 |
4.29 |
The P/Cr and O/P atomic ratios in the chromium phosphates synthesized via the SSRLT route were determined via EDS, and the results are listed in Table 1. One can see that the P/Cr atomic ratio in the synthesized samples is approximately equal to those in the initial batches, and the O/P atomic ratio is around 4. It is clear that the chemical compositions of synthesized chromium phosphates correlate well with the theoretical values.
Fig. 2 presents the HR-TEM micrograph of a mesoporous chromium phosphate synthesized via the SSRLT route from an initial batch with the atomic ratio P/Cr = 2.0. It can be seen that the sample exhibits an orderly wormhole-like pore array structure before the surfactant removal (Fig. 2a) and a banger-like pore geometry after the surfactant removal (Fig. 2b). The average distance between the centers of mesopores, observed from the micrographs, is ca. 4 nm, which correlates well with the result from the XRD datum. The formation of the mesostructure in the chromium phosphate is probably due to the surfactant CTAB and chromium phosphate forming a uniform and orderly solid array, which consists of the rod-like micelles of the CTAB surrounded by chromium phosphates,37 as shown in Fig. 2a. Then, a uniform and orderly pore array structure is formed when the rod-like micelles of the CTAB are removed from this solid array through calcination (Fig. 2b). Thus, the surfactant CTAB plays an important role in the mesopore structure of chromium phosphates.
 |
| | Fig. 2
HR-TEM
micrograph of a mesoporous chromium phosphate prepared from an initial batch with an atomic ratio of P/Cr=2.0 before (a) and after (b) the surfactant removal. | |
The nitrogen adsorption–desorption isotherm and the pore size distribution curve (in the inset) of the mesoporous chromium phosphate synthesized via the SSRLT route (with P/Cr atomic ratio = 2.0) are shown in Fig. 3. The isotherm can be ascribed to a type IV in the IUPAC classification and indicates a typical mesostructure. The desorption hysteresis loop corresponds to a type H2, and is caused by pore blocking effects during desorption,38 indicating an ink-bottle pore geometry of the mesostructure. From Fig. 3, a sharp increase of the volume adsorbed at very low relative pressures (P/P0) is observed, indicating that the walls of the mesopores contain micropores. The BET specific surface area is 250.78 m2g−1 and the pore volume 0.21 cm3g−1, which have been calculated on the basis of adsorption in the relative pressure range from 0.05 to 0.2 and at a relative pressure of 0.9814, respectively. The mesoporous chromium phosphate has a uniform and narrow pore size distribution, with most of the pore sizes centering around 3.48 nm. Tarafdar et al34 synthesized the mesoporous chromium phosphate from an initial batch with an atomic ratio P/Cr = 1.0 at 353–393 K, via the solution sol–gel route. The obtained sample had a specific surface area of ca. 384 m2 g-1 and a pore volume of 0.2937 m3 g-1. We also synthesized the mesoporous chromium phosphate from an initial batch with an atomic ratio P/Cr = 2.0, according to Tarafdar’s method, and the obtained sample possessed a specific surface area of 213.30 m2 g-1 and an average pore size of 3.10 nm. However, when the mesoporous chromium phosphate samples, synthesized via the solution sol-gel, were heated at 823 K for 4 h, the specific surface area decreased rapidly to 201 m2 g−1 due to the thermal-collapse of the mesostructure. In this SSRLT work, the mesoporous chromium phosphate was calcined at 823 K for 6 h to remove the surfactant before the N2-physisorption measurement. Therefore, it is reasonable to conclude that the mesoporous chromium phosphate synthesized via the SSRLT route possesses a higher specific surface area and more thermal stability than that prepared via the solution sol–gel route
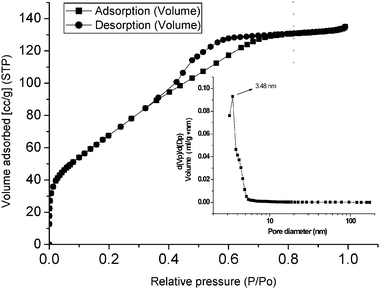 |
| | Fig. 3 Adsorption–desorption isotherm and pore size distribution curve (inset) of a mesoporous chromium phosphate prepared from an initial batch with an atomic ratio of P/Cr = 2.0. | |
The result of the thermal analysis on the mesoporous chromium phosphate synthesized via the SSRLT route (with P/Cr atomic ratio = 2 and without the surfactant removal) is shown in Fig. 4. The weight loss amounting to 6.5 wt% in the TG curve below 423 K is due to dehydration, and the small and broad endothermic band is present in the DSC curve in this temperature range. From 423–683 K, a weight loss of 24.5 wt% is observed in the TG curve, and an endothermic peak centered at 603 K is present in the DSC curve. This peak can be ascribed to the decomposition of the long-chain alkyl group of the surfactant CTA cations. A weight loss of 14.0 wt% is observed in the temperature range 683–793 K in the TG curve, and correspondingly, an endothermic peak centered at 733 K in the DSC curve is present. This peak is attributed to the decomposition of the head-group of the surfactant CTA cations. For temperatures higher than 793 K, a weigh loss of as small as 2.0 wt% is observed in the TG curve along with a very broad exothermic band in the DSC curve. This may be due to the gradual phase transformation of the mesoporous chromium phosphate from ordered mesophase to distorted mesophase at temperatures above 793 K.
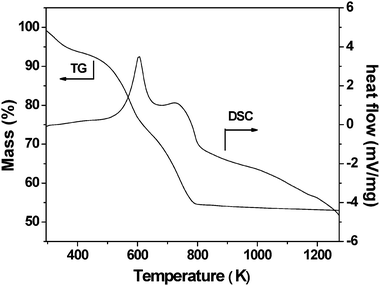 |
| | Fig. 4
TG-DSC curves for the mesoporous chromium phosphate synthesized via the SSRLT route from an initial batch with an atomic ratio of P/Cr = 2.0. | |
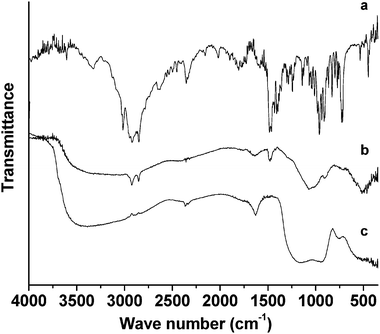
The FT-IR spectra of CTAB and mesoporous chromium phosphate synthesized via the SSRLT route (with atomic ratio P/Cr = 2.0) is shown in Fig. 5. The spectrum of the pure surfactant CTAB (Fig. 5a) exhibits the characteristic vibration absorptions of a quaternary ammonium compound with a long alkyl chain, i.e., ν(CH3) and ν(CH2) (3020, 2920, and 2850 cm−1), δ(CH3) and δ(CH2) (1480–1360 cm−1), ν(C–N) (1290 and 1240 cm−1), νas(N–R4) (∼960 cm−1), γ(CH2) (730 cm−1), and νs(N–R4) (∼720 cm−1).39 With the exception of the band at 2347 cm−1 (νas(CO2)),40 the weak bands in the range 2650–1600 cm−1 probably reflect combinations or overtones of the skeleton of the CTAB. In the spectrum of mesoporous chromium phosphate containing the CTAB (Fig. 5b), some of the characteristic vibration bands of the CTAB (2920, 2850, 1480, 1290, 910, and 715 cm−1) are presented. Besides several broad bands centered at 3500 cm−1 (ν(OH or H3O+)34,41–44 and/or ν(P–OH)41), those at 1630 cm−1 (δas(OH or H3O+),23,41,43 1070 cm−1 (νs(PO4)),1,10,23,41–44 and 505 cm−1 (δout(POH)40 and/or δ(PO4)23,41,43) are also detected. Compared with Fig. 5a, the intensities of the absorption bands of both the alkyls and the N–R4 head group in the CTAB in Fig. 5b are much smaller, especially for the N–R4 head group. The wave numbers of the absorption bands of the N–R4 head group shifted from 960 and 720 cm−1 in Fig. 5a to 910 and 715 cm−1 in Fig. 5b. These results indicate that the CTAB has been incorporated into the mesopores of chromium phosphate, and there is a strong interaction between the head group of the CTAB and the moieties of chromium phosphate, which contributes to the building of the mesoporous chromium phosphate. In the spectrum of mesoporous chromium phosphate after the CTAB removal (Fig. 5c), all the bands of the CTAB disappear except those of the phosphates (shown in Fig. 5b), which remain. Two new bands (930 and 755 cm−1) appear (Fig. 5c) due to the mixed motions involving both the PO4 tetrahedron and the CrO6 octahedron.10,44 The characteristic vibrations due to the skeleton of PO4 (νs(PO4) and δ(PO4)) shift from 1070 and 505 cm−1, for the sample containing the surfactant CTAB, to 1130 and 530 cm−1, for the sample without the surfactant CTAB. This indicates that, in the formation of mesoporous chromium phosphates, the phosphates play an important role by interacting first with the surfactant CTAB, thus stimulating the assembly of chromium ions around the surfactant molecules. There is a stronger interaction between the chromium and the phosphate ions after the surfactant removal.
Fig. 6 shows the FT-IR spectra of the chromium phosphate samples prepared from the initial batches with various P/Cr atomic ratios before the surfactant removal. For P/Cr ≥ 2.0 (Fig. 6d–g), the spectra are very similar to each other and the vibration band assignment is similar to that of P/Cr = 2.0 in Fig. 5b. The spectrum for P/Cr = 1.8 (Fig. 6c) is similar to that of P/Cr ≥ 2.0, but the intensity of the band at 2347 cm−1 (νas(CO2)) is much larger in the former than that in the latter. For P/Cr < 1.8 (Fig. 6a–b), most of the vibration bands observed for P/Cr = 1.8 in Fig. 6c are nearly unchanged except for the absence of the vibration bands of the CTAB.
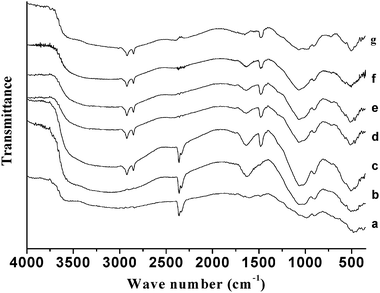 |
| | Fig. 6
FT-IR spectra of mesoporous chromium phosphate prepared from initial batches with various P/Cr atomic ratios before the surfactant removal. The P/Cr atomic ratio is equal to (a) 1.0, (b) 1.6, (c) 1.8, (d) 2.0, (e) 2.2, (f) 2.4; and (g) 3.0. | |
Fig. 7 presents the FT-IR spectra of chromium phosphate samples prepared from initial batches with various P/Cr atomic ratios after the surfactant removal. The spectra are similar to those of Fig. 5c; however, the intensity of the band at 2347 cm−1 (νas(CO2)) is obviously larger for P/Cr ≤ 1.8 (Fig. 7a–c) than for P/Cr > 1.8 (Fig. 7d–g).
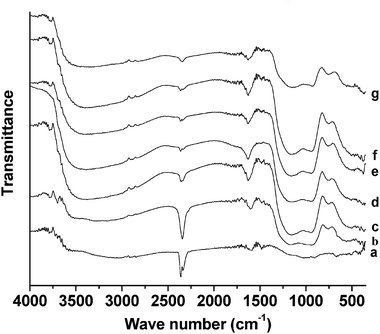 |
| | Fig. 7
FT-IR spectra of mesoporous chromium phosphate prepared from initial batches with various P/Cr atomic ratios after the surfactant removal. The P/Cr atomic ratio is equal to (a) 1.0, (b) 1.8, (c) 2.0, (d) 2.0, (e) 2.2, (f) 2.4, and (g) 3.0. | |
The FT-IR spectra in Fig. 5–7 show that no CTAB was incorporated into the chromium phosphate when the samples were prepared from the initial batches with atomic ratios of P/Cr < 1.8. From the XRD characterization of these samples, we found that chromium phosphate mesophases were formed only for the atomic ratios where P/Cr ≥ 1.8 (seen in Fig. 1). Therefore, the surfactant CTAB plays a key role in the formation of the mesoporous chromium phosphate. It probably involves the formation of mesoporous chromium phosphate by first assembling the phosphates around the head groups of the surfactant CTAB and, subsequently, assembling chromium around the moiety of the CTAB and the phosphates. The removal of the surfactant promotes the interaction between the phosphate and the chromium ions, resulting in the formation of chromium phosphate. The higher P/Cr atomic ratios (>1.8) facilitate the interactions of the CTAB and the phosphates, which in turn, results in the incorporation of the surfactant CTAB into the chromium phosphates. Another result of the higher atomic ratios of P/Cr (>1.8) is that the chromium ions are surrounded by a larger number of phosphates; thus, the adsorption of CO2 over chromium is largely hindered, which is in contrast to the case with the lower atomic ratios of P/Cr (<1.8). The evidence of this is the larger intensity of the CO2 vibration bands for P/Cr < 1.8 than for P/Cr > 1.8 (cf.Fig. 6a–c and d–g and 7a–c and d–g).
Fig. 8 presents the DR UV-Vis spectrum of a mesoporous chromium phosphate prepared from an initial batch with an atomic ratio of P/Cr = 2.0 after the removal of CTAB. Having a d3 electronic configuration, the Cr(III) ion usually has an octahedral coordination, due to its high crystal field stabilization energy.45 Theoretically, the DR UV-Vis spectrum of the Cr(III) ion in the octahedral crystal field exhibits mainly three- spin-allowed d–d electron transitions 4A2g(F) → 4T1g(P), 4A2g(F) → 4T1g(F), and 4A2g(F) → 4T2g(F) at 290, 450, and 664 nm, respectively, and several spin-forbidden transitions.46,47 Ravikumar et al.48 detected the above three spin-allowed transitions at 290, 450, and 664 nm in the UV-DR spectrum of Cr(III) doped on a zinc phosphate glass substrate. In addition, the 664 nm band exhibited two dips as a fine structure at ca. 650 and 687 nm, due to the spin-forbidden transitions 4A2g(F) → 2E2g(G) and 4A2g(F) → 2T1g(G). Depending on the substrate and/or coordination environment in which Cr(III) ions are located, red- or blue-shifts can occur for the above bands, and only some of the above bands could be detected in the chromium-containing compounds.6,41,49-60 In Fig. 8, all the above five bands at 290, 445, 640, 665, and 688 nm have been detected, and they can be ascribed to the d–d electron transitions from 4A2g (F) to T1g (P), 4T1g (F), 4T2g (F), 2E2g (G), and 2T1g (G), respectively. This suggests that, in the mesoporous chromium phosphate prepared via the SSRLT route, the Cr(III) ions are highly dispersed and have an octahedral coordination. EDS has revealed that the sample has atomic ratios of P/Cr = 2.0 and O/P = 4.69 (Table 1). One can conclude that two phosphates (providing two oxygen coordinates per phosphate) and 1–2 water molecules (providing one oxygen coordinate per water molecule) generate an octahedral coordination environment around the Cr(III) ion in the mesoporous chromium phosphate. The bands at 420 and 480 nm, as shoulders of the band 4A2g (F) → 4T1g (F), are probably caused by the different micro-environments around the Cr(III) ions in the mesoporous chromium phosphate. The slight distortion of the octahedral coordination, due to the partial loss of water molecules around some Cr(III) ions, might be a possible cause. In addition, a strong band at 215 nm and a weak band at 335 nm are identified in Fig. 8. The strong bands at ca. 270 and 340 nm are usually attributed to an O → Cr(VI) charge transfer and/or to a polychromate species.6,48,49,51,53,54,56,58,59,61,62 Because the band at ca. 270 nm could not be detected, and that at 335 nm is very weak (Fig. 8), the content of the Cr(VI) ions and/or the polychromate species in the mesoporous chromium phosphate is expected to be low. It is well known that the oxidized states Cr(IV) and Cr(V) are rather unstable, and Cr(V) can be easily disproportionate to Cr(III) and Cr(VI). In addition, Cr(II) ions are strong reducing agents and are stable only in the absence of oxygen. Therefore, it is reasonable for us to assign the bands at 210 nm and 335 nm to the O → Cr(III) charge transfer.
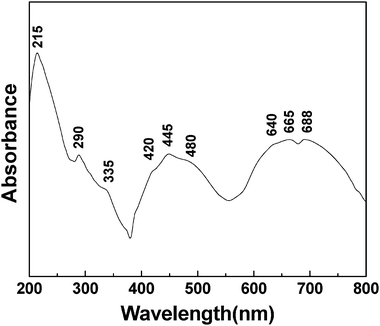 |
| | Fig. 8 The UV-DR spectrum of a mesoporous chromium phosphate prepared from an initial batch with an atomic ratio of P/Cr = 2.0 after the surfactant removal. | |
The α-CrPO4 possesses a structure of infinite network constructed by apically linking [CrO6] octahedra to the pairs of edge-sharing [CrO6] octahedra, with PO4 tetrahedra being linked to the network, while the structure of β-CrPO4 contains infinite chains of trans edge-sharing [CrO6] octahedra linked together by [PO4] tetrahedra.63,64 In contrast to the crystalline CrPO4, the amorphous CrPO4 consists of short chains of [CrO6] octahedra with finite and irregular lengths (defect structures).6 We identified that the walls of the mesoporous chromium phosphates prepared via the SSRLT route possess amorphous structures, which might be somewhat different from those reported in the literature.6,63,64 The XRD and FT-IR experiments have shown that mesophases are formed only for atomic ratios P/Cr ≥ 1.8, and the DR UV-Vis spectrum reveals that chromium ions are present mainly as Cr(III) in octahedral coordination surrounded by phosphates and water molecules. EDS data have shown that the P/Cr atomic ratios of the samples are nearly equal to those in the initial batches, and the O/P atomic ratios for all the samples are around 4 (see Table 1). It suggests that both the chromium and the phosphate are not in an oligomerized state; every chromium ion is octahedrally coordinated with the oxygen atoms from the phosphates and the water molecules, thus forming the chromium phosphate moiety. In this way, the Cr(III) ions are well separated by phosphates and are highly dispersed in the phosphate matrix. This is important for the catalytic applications of mesoporous chromium phosphates because a high dispersion of the active phase and/or active centers normally results in a higher performance of a heterogeneous catalyst.
Based on the data from XRD, FT-IR, and UV-DR, a mechanism of the formation of the chromium phosphate mesophase is proposed. Because hydrates of chromium and phosphate are used as precursors in the synthesis, water molecules released from the hydrates function as the “reagent” rather than the “solvent,” which promotes homogeneously mixing the CTAB, chromium, and phosphate by grinding. The aggregates of the CTAB function as the templates to guide the construction of the mesophase pore structure. Through ionic exchange between the phosphate and the bromide of the CTAB, the phosphate ions interact first with the head groups of CTAB and then connect with the chromium ions. As in conventional solid state reaction, the double decomposition reaction and ion diffusion constitute the steps for the formation of the walls of the mesopores.
Table 2 shows the effect of reaction temperature on isopropanol dehydration to propene over the mesoporous chromium phosphate catalyst synthesized via the SSRLT route from an initial batch with an atomic ratio P/Cr = 2.0. One can see that the isopropanol conversion increases but the propene selectivity exhibits a maximum at 533 K, with the increase of reaction temperature, and the largest yield of propene (89.78%) is achieved at 533 K. It was reported in ref. 4 that, over a non-mesoporous chromium phosphate catalystvia the conventional sol-gel route from an initial batch with an atomic ratio P/Cr = 1.0, both the isopropanol conversion and propene selectivity increased with the increase of reaction temperature (in the range 475–575 K), providing a maximum yield of propene (ca. 35%) at 575 K.4 Thus, a higher yield of propene at lower reaction temperature can be obtained over the mesoporous catalyst than over the non-mesoporous one.
Table 2 Effects of reaction temperature on isopropanol dehydration to propene over mesoporous chromium phosphate catalysts synthesized via the SSRLT route from an initial batch with an atomic ratio of P/Cr = 2.0
|
T/K |
X (%) |
S
i
(%) |
Y (%) |
|
Propene
|
Diisopropyl ether
|
| Note: X = conversion of isopropanol; Si, selectivity to component i; Y is the yield of propene. |
| 473 |
33.14 |
71.34 |
28.66 |
23.64 |
| 503 |
58.97 |
94.66 |
5.34 |
55.82 |
| 533 |
93.10 |
96.43 |
3.57 |
89.78 |
| 563 |
96.37 |
86.33 |
13.67 |
83.19 |
Table 3 presents the results of isopropanol dehydration to propene at 533 K over the chromium phosphate catalysts synthesized via the SSRLT and sol-gel routes. The mesoporous chromium phosphate catalyst synthesized via the SSRLT route from an initial batch with an atomic ratio P/Cr = 1.0 provides a propene selectivity of 90.74% at an isopropanol conversion of 50.49%. This result is superior to that reported in Ref. 4, where a propene selectivity of ca. 86% and an isopropanol conversion of ca. 15% at 533 K was obtained over the non-mesoporous chromium phosphate catalyst. By increasing the P/Cr atomic ratio in the initial batch, both the isopropanol conversion and the propene selectivity first increase and then decrease over the mesoporous chromium phosphate catalyst synthesized via the SSRLT route. The highest propene selectivity (96.43%) and isopropanol conversion (93.10%) are achieved over the catalyst with an atomic ratio of P/Cr = 2.0 in the initial batch. As comparisons, a propene selectivity of 94.33% at an isopropanol conversion of 68.89% is obtained over the mesoporous chromium phosphate synthesized via the solution sol–gel route from an initial batch with an atomic ratio of P/Cr = 2.0, whereas a propene selectivity of 88.05% at an isopropanol conversion of 51.84% is achieved over the non-mesoporous chromium phosphate catalyst synthesized via the conventional sol-gel route from an initial batch with an atomic ratio of P/Cr = 2.0. The above results indicate that, for the dehydration of isopropanol to propene, the mesoporous catalysts are superior to the non-mesoporous one; furthermore, the mesoporous catalyst synthesized via the RSSLT route has a much better performance than that synthesized via the solution sol-gel route.
Table 3 Dehydration of isopropanol to propene over chromium phosphate catalysts synthesized via the SSRLT and sol–gel routes. (Reaction temperature 533 K)
|
Catalysts
|
X (%) |
S (%) |
Y (%) |
| Synthesis route |
P/Cr molar ratio in initial batch |
Propene
|
Diisopropyl ether
|
| Note: X = conversion of isopropanol; Si = selectivity to component i; Y = yield of propene. Non-mesoporous chromium phosphate; Mesoporous chromium phosphate. |
|
SSRLT
|
1.00 |
50.49 |
90.74 |
9.26 |
45.82 |
|
SSRLT
|
1.60 |
67.82 |
95.67 |
4.33 |
64.88 |
|
SSRLT
|
1.80 |
72.45 |
96.08 |
3.92 |
69.60 |
|
SSRLT
|
2.00 |
93.10 |
96.43 |
3.57 |
89.78 |
|
SSRLT
|
2.20 |
92.43 |
96.06 |
3.94 |
88.79 |
|
SSRLT
|
2.40 |
91.34 |
95.85 |
4.15 |
87.55 |
| Sol–gela |
2.00 |
51.84 |
88.05 |
11.95 |
45.64 |
| Sol–gelb |
2.00 |
68.89 |
94.33 |
5.67 |
64.98 |
It has been shown in Section 3.1 that in the synthesized mesoporous chromium phosphates, the O/P atomic ratio is around 4.0 and the P/Cr atomic ratio is very close to that in the initial batches. The chromium phosphates with an atomic ratio P/Cr ≥ 1.8 possess a mesostructure, with the highest crystallinity of mesophase appearing at P/Cr = 2.0, while those with atomic ratio P/Cr < 1.8 are non-mesoporous. The BET specific surface area and average pore size (250.78 m2g−1 and 3.48 nm) for the mesoporous chromium phosphate synthesized via the RSSLT route are much larger than those (213.30 m2g-1 and 3.10 nm) for the mesoporous chromium phosphate synthesized via the sol-gel route. Therefore, the mesostructure of chromium phosphate catalyst contributes in an important way to its catalytic performance in the dehydration of isopropanol to propene, mostly due to the high crystallinity, high surface area, high active centers/phase, and accessibility of the active sites of the catalyst. The dehydration of alcohols is usually catalyzed by the acidic sites located on heterogeneous catalysts.4 The surface acidities of chromium phosphate catalysts were found to be affected largely by the preparation method.4,7,9,10 The increase in the calcination temperature of the chromium phosphate catalyst prepared via the conventional sol-gel method caused the decrease in the catalytic activity of dehydration of isopropanol to propene, due to the decrease in surface acidity.4 One can expect that the performances for the dehydration of isopropanol to propene over the mesoporous chromium phosphate catalysts synthesized in this work are related to their acidic character, which are affected by the P/Cr atomic ratios in the catalysts. The results in section 3.1 have indicated that in the mesoporous chromium phosphate synthesized via the SSRLT route, both the chromium and the phosphate are not in an oligomerized state, and every chromium ion is octahedrally coordinated by both phosphates and water. A potential unsaturation of coordination around the chromium would be introduced, with the water molecules being lost during catalytic reaction. Thus, the isopropanol molecules may be coordinated to chromium and catalytically dehydrated by the phosphate. With the increase of the P/Cr atomic ratio, the acidity of the catalyst increases, but its potential unsaturation of coordination decreases, resulting in an optimized catalytic performance at an atomic ratio of P/Cr = 2.0. Overall, for mesoporous chromium phosphates, both the P/Cr atomic ratio and mesostructure play an important role in the catalytic performance.
4. Conclusion
A novel method that employs both the solid-state reaction at low temperature (SSRLT) and surfactant CTAB, for the synthesis of mesoporous chromium phosphate, has been successfully developed. The mesoporous chromium phosphate synthesized possessed a specific surface area of as high as 250.78 m2 g−1 and a narrow pore size distribution. The walls of mesopores had an amorphous structure and consisted of highly-dispersed chromium (III) surrounded octahedrally by phosphates. The formation of the mesoporous chromium phosphate was a result of supramolecular assembly, with the aggregates of surfactant molecule CTAB acting as the structure-directing template, which involves the interaction of phosphate ions with the head groups of CTAB as the key step. The mesoporous chromium phosphate catalyst synthesized via the SSRLT route exhibited a significantly higher catalytic performance in the dehydration of isopropanol to propene, as compared with the amorphous chromium phosphate catalyst synthesized via the solution sol-gel route and the non-mesoporous chromium phosphate. Over the mesoporous chromium phosphate catalyst synthesized via the SSRLT route from an initial batch with an atomic ratio P/Cr = 2.0, a 93.10 % conversion of isopropanol at a 96.43 selectivity to propene have been achieved.
Appendix
The dehydration of isopropanol to propene, the conversion of isopropanol, the selectivity to propene, and the yield of propene were calculated as follws:
| Selectivity to component i: Si (%) = (γi × Ni)/(∑γi × Ni) |
where
| Ni = V × Ci,g/22400 + W × Ci,l/Mi; |
N
i
: total amount of component i in gaseous and liquidic products, mol; V: flow rate of gaseous products, ml min−1; W: weight of liquid products collected in unit time, g min−1; Ci,g: volume concentration of component i in gaseous products, %; Ci,l: weight concentration of component i in liquid products, %; Mi: molar weight of component i in liquid products; γi: ratio of carbon numbers in component i to that in isopropanol; Niso: total amount of isopropanol in gaseous and liquid products, mol.
Acknowledgements
This work was supported by the State Key Laboratory of Chemo/Biosensing and Chemometrics, China, and the Program for Lotus Scholar in Hunan Province, China.
References
- P. Yin, Y. Hu, Y. Sun, Y. Yang, C. N. Ji, Q. Xu, X. H. Xu and W. J. Zhang, Mater. Lett., 2007, 61, 3755 Search PubMed.
- S. Mustafa, S. Murtaza, A. Naeem and K. Farina, J. Colloid Interface Sci., 2005, 283, 287 Search PubMed.
-
M. M. Johnson and G. P. Nowack, U. S. Patent, 4558165, 1985 Search PubMed.
- F. M. Bautista, J. M. Campelo, A. Garcia, D. Luna, J. M. Marinas, A. A. Romero and M. R. Urbano, React. Kinet. Catal. Lett., 1995, 55, 133 Search PubMed.
- Y. C. Kuo and Y. W. Chen, J. Chin. Inst. Chem. Eng., 2000, 31, 1 Search PubMed.
- M. Loukah, G. Coudurier, J. C. Vedrine and M. Ziyad, Microporous Mater., 1995, 4, 345 Search PubMed.
- F. M. Bautista, J. M. Campelo, A. Garcia, D. Luna, J. M. Marinas, A. A. Romero and M. R. Urbano, React. Kinet. Catal. Lett., 1994, 53, 55 CAS.
- F. M. Bautista, J. M. Campelo, A. Garcia, D. Luna, J. M. Marinas, A. A. Romero and M. R. Urbano, React. Kinet. Catal. Lett., 1998, 64, 41 Search PubMed.
- F. M. Bautista, J. M. Campelo, A. Garcia, D. Luna, J. M. Marinas, A. A. Romero and M. R. Urbano, React. Kinet. Catal. Lett., 1997, 62, 47 Search PubMed.
- F. M. Bautista, J. M. Campelo, A. Garcia, D. Luna, J. M. Marinas and M. R. Urbano, React. Kinet. Catal. Lett., 1993, 49, 173 CAS.
- N. Stock, G. Ferey and A. K. Cheetham, Solid State Sci., 2000, 2, 307 Search PubMed.
- E. A. Merritt, M. Sundaralingam and D. Dunaway-Mariano, J. Am. Chem. Soc., 1981, 103, 3565 CrossRef CAS.
- E. Rodek, W. Sterzel and N. Theile, Z. Anorg. Allg. Chem., 1980, 462, 42 Search PubMed.
- J. P. Attfield, A. W. Sleight and A. K. Cheetham, Nature, 1986, 322, 620 CrossRef CAS.
- R. Glaum and R. Gruehn, Z. Anorg. Allg. Chem., 1986, 543, 111 Search PubMed.
- J. P. Attfield, P. D. Battle and A. K. Cheetham, J. Solid State Chem., 1985, 57, 357 CAS.
- M. Gruss and R. Glaum, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1996, 52, 2647 CrossRef.
- R. Glaum and A. Schmidt, Z. Anorg. Allg. Chem., 1997, 623, 1672 Search PubMed.
- R. Glaum, M. Walter-Peter, D. Ozalp and R. Gruehn, Z. Anorg. Allg. Chem., 1991, 601, 145 Search PubMed.
- R. Glaum, Z. Kristallogr., 1993, 205, 69 Search PubMed.
- R. Glaum, Z. Anorg. Allg. Chem., 1992, 616, 46 Search PubMed.
- W. Liu, D. B. Xiong, X. X. Yang and J. T. Zhao, J. Solid State Chem., 2007, 180, 2116 CrossRef CAS.
- C. T. Kresge, M. E. Leonowicz, W. J. Roth, J. C. Vartuli and J. S. Beck, Nature, 1992, 359, 710 CrossRef CAS.
- D. Y. Zhao, J. L. Feng, Q. S. Huo, N. Melosh, G. H. Fredrickson, B. F. Chmelka and G. D. Stucky, Science, 1998, 279, 548 CrossRef CAS.
- S. S. Kim, W. Zhang and T. J. Pinnavaia, Science, 1998, 282, 1302 CrossRef CAS.
- P. T. Tanev and T. J. Pinnavaia, Science, 1995, 267, 865 CrossRef CAS.
- D. M. Antonelli and J. Y. Ying, Angew. Chem., Int. Ed. Engl., 1996, 35, 426 CrossRef CAS.
- P. Yang, D. Zhao, D. Margolese, B. F. Chmelka and G. D. Stucky, Nature, 1998, 396, 152 CrossRef CAS.
- A. C. Vosloo, Fuel Process. Technol., 2001, 71, 149 CrossRef CAS.
- E. Ruckenstein and Z. S. Chao, Nano Lett., 2001, 1, 739 Search PubMed.
- J. J. Shyue and M. R. DeGuire, J. Am. Chem. Soc., 2005, 127, 12736 Search PubMed.
- G. Wu, X. Wang, B. Chen, J. Li, N. Zhao, W. Wei and Y. Sun, Appl. Catal., A, 2007, 329, 106 Search PubMed.
- T. Kimura, Microporous Mesoporous Mater., 2005, 77, 97 CrossRef CAS.
- A. Tarafdar, S. Biswas, N. K. Pramanik and P. Pramanik, Microporous Mesoporous Mater., 2006, 89, 204 Search PubMed.
- X. M. Liu, G. Q. Lu and Z. F. Yan, J. Phys. Chem. B, 2004, 108, 15523 CrossRef CAS.
- M. Thommes, Chem. Ing. Tech., 2010, 82, 1059 CrossRef.
- W. Li, M. Zhang and J. L. Zhang,
et al.
, Front. Chem. China, 2006, 1, 438 Search PubMed.
-
S. J. Gregg and K. S. W. Sing, Adsorption, Surface Area and Porosity, Academic Press, London, 1982 Search PubMed.
-
John A. Dean, Analytic Chemistry Handbook, McGraw-Hill, 1995 Search PubMed.
- M. Taraschewski, H. K. Cammenga, R. Tuckermann and S. Bauerecker, J. Phys. Chem. A, 2005, 109, 3337 CrossRef CAS.
- C. M. D. O. Raposo and J. G. Eon, Mater. Res., 2002, 5, 421 Search PubMed.
- M. Wakamura, K. Kandori and T. Ishikawa, Polyhedron, 1997, 16, 2047 Search PubMed.
- F. M. Bautista, J. M. Campelo, A. Garcia, D. Luna, J. M. Marinas and M. R. Urbano, J. Mater. Chem., 1994, 4, 311 RSC.
- A. Q. Yuan, S. Liao, Z. F. Tong, J. Wu and Z. Y. Huang, Mater. Lett., 2006, 60, 2110 Search PubMed.
-
A. F. Wells, Structural Inorganic Chemistry, Clarendon Press, Oxford, 1984 Search PubMed.
-
C. J. Ballhausen, Introduction to Ligand Field Theory, McGraw-Hill Book Co., New York, 1962 Search PubMed.
-
A. B. P. Lever, Inorganic Electronic Spectroscopy, Elsevier Pub. Co., Amsterdam, 1984 Search PubMed.
- R. V. S. S. N. Ravikumar, J. Yamauchi, A. V. Chandrasekhar, Y. P. Reddy and P. Sambasiva Rao, J. Mol. Struct., 2005, 740, 169 Search PubMed.
- L. C. Liu, H. Q. Li and Y. Zhang, J. Phys. Chem. B, 2006, 110, 15478 Search PubMed.
- L. Frunza, J. Pelgrims, H. Leeman, P. Van Der Voort, E. F. Vansant, Robert, A. Schoonheydt and B. M. Weckhuysen, J. Phys. Chem. B, 2001, 105, 2677 CrossRef CAS.
- M. Hartmann, Chem. Rev., 1999, 99, 635 CrossRef CAS.
- A. M. Beale, D. Grandjean, J. Kornatowski, P. Glatzel, F. M. F. de Groot and B. M. Weckhuysen, J. Phys. Chem.
B, 2006, 110, 716 CrossRef CAS.
- S. C. Laha, G. Kamalakar and R. Glaser, Microporous Mesoporous Mater., 2006, 90, 45 CrossRef CAS.
- S. C. Laha and R. Glaser, Microporous Mesoporous Mater., 2007, 99, 159 Search PubMed.
- S. Shylesh, Ch. Srilakshmi, A. P. Singh and B. G. Anderson, Microporous Mesoporous Mater., 2007, 99, 334 CrossRef CAS.
- S. K. Mohapatra, F. Hussain and P. Selvam, Catal. Lett., 2003, 85, 217 CrossRef CAS.
- M. Miyake, H. Uehara, H. Suzuki, Z. D. Yao, M. Matsuda and M. Sato, Microporous Mesoporous Mater., 1999, 32, 45 Search PubMed.
- Ch. Subrahmanyam, B. Louis, F. Rainone, B. Viswanathan, A. Renken and T. K. Varadarajan, Appl. Catal., A, 2003, 241, 205 CrossRef CAS.
- Ch. Subrahmanyam, B. Louis, F. Rainone, B. Viswanathan, A. Renken and T. K. Varadarajan, Catal. Commun., 2002, 3, 45 Search PubMed.
- B. M. Weckhuysen, I. E. Wachs and R. A. Schoonheydt, Chem. Rev., 1996, 96, 3327 CrossRef CAS.
- R. K. Jha, S. Shylesh, S. S. Bhoware and A. P. Singh, Microporous Mesoporous Mater., 2006, 95, 154 CrossRef CAS.
- J. D. Chen and R. A. Sheldon, J. Catal., 1995, 153, 1 CrossRef CAS.
- J. P. Attfield, P. D. Battle and A. K. Cheetham, J. Solid State Chem., 1985, 57, 357 CAS.
- J. P. Attfield, P. D. Battle, A. K. Cheetham and D. C. Johnson, Inorg. Chem., 1989, 28, 1207 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2012 |
Click here to see how this site uses Cookies. View our privacy policy here.