The influence of ultrasound on porphyrin-based metallogel formation: efficient control of H- and J-type aggregations†
Youngje
Cho
a,
Ji Ha
Lee
a,
Justyn
Jaworski
ab,
Sunhong
Park
a,
Shim Sung
Lee
a and
Jong Hwa
Jung
*a
aDepartment of Chemistry and Research Institute of Natural Science, Gyeongsang National University, Jinju 660-701, Korea. E-mail: jonghwa@gnu.ac.kr
bDepartment of Chemical Engineering, Hanyang University, Seoul 133-791, Korea
First published on 3rd November 2011
Abstract
Efficient control of aggregation modes of the porphyrin–Pd2+ complexes by ultrasonication and heating is reported. A porphyrin-based metallogel is formed in the presence of Pd2+, which displays H-type aggregation. Heating the porphyrin derivative in the presence of Pd2+, however, results in J-type aggregation that does not produce a gel. Conclusive evidence was found that the crystal structure of the related porphyrin–Pd2+ complex obtained by heating shows J-type aggregation.
Stimuli-responsive aggregations of low molecular weight materials have been intensively studied in gels, micelles, vesicles and synthetic membranes with the aim of creating future technologies for the control of fluidity, viscoelasticity, solvent volatility, optical transmission and ion transport, etc.1 Light2 and electrochemical controls3 have been explored as a means to switch on molecular aggregations without the use of chemical stimuli. However, devising a method of control that is instantaneous, positive (i.e., converting sol to aggregate), and reversible, while also being versatile and practical, remains a challenge.
In general, supramolecular gels are prepared by heating a gelator in an appropriate solvent, followed by cooling the resulting isotropic supersaturated solution to room temperature.4 In contrast, recently, Naota and Koori reported metallogel formation by ultrasonication.5 The employment of sonication as an unexpected but effective stimulus to promote hydrogen bonding, π–π stacking and/or van der Waals interaction-dependent molecular gel formation has received unprecedented attention. However, ultrasound-controlled supramolecular gel formation based on metal coordination interactions has been less explored, and to the best of our knowledge, only a few examples have been described so far.5,6
Since porphyrins tend to align into one-dimensional aggregates, they are of much interest in relation to the creation of novel supramolecular architectures such as nanowires, discotic liquid crystals, helical ribbon structures, etc. The aggregation models of the porphyrin-based organogelators are classified into two types, H- and J-aggregates.7–10 Both aggregation models are closely related to intermolecular hydrogen-bonding interactions by the edge-to-edge interaction mode of the porphyrin planes. Nonetheless, it has been difficult to establish a simplified correlation between structure and aggregation modes from a library of porphyrin derivatives.11–13 In addition, no reports on controlled aggregation models of porphyrin-based gelators have appeared. Here, we report that the H- and J-aggregation modes of metal-based porphyrin gelators can be controlled by ultrasonication and heating. This is the first example of controlled aggregation models using these different methods in the formation of metallogels.
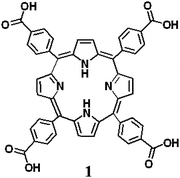
When a homogeneous, clear solution of 1 in the presence of Pd2+ in various organic solvents was irradiated with ultrasound for a few seconds, the stable sol state was completely converted to a gel state immediately after sonication. Typically, ultrasonication (0.45 W cm−2, 40 kHz) of 7.6 × 10−5 M solution of 1 in the presence of Pd2+ (1.0 equiv.) in acetonitrile for 10 s at 293 K gave an entirely opaque gel (Fig. 1a). The gel formation of 1 in the presence of Pd2+ might be attributed to the π–π stacking between porphyrin and porphyrin moieties, due to enhancement of the translational motion by ultrasonication. Without ultrasonication, heating the solution of 1 with Pd2+ under the same conditions did not cause gel formation (Fig. 1b). The gel that was formed by ultrasonication was stable, but it was readily converted to a sol by heating above the gelation temperature, Tgel, and subsequently cooling to room temperature. The present sol–gel phase transition, initiated by an external stimulus, can be repeated indefinitely with no measurable degradation of 1. The gelation of the porphyrin-based metallogel occurs exclusively upon ultrasonication, since other external stimuli such as vigorous shaking, quick heating/cooling and microwave irradiation do not initiate gel formation. Porphyrin derivative 1 without metal ions did not form a gel upon ultrasonication or heating when used with organic solvents, which is a factor of solubility.
 | ||
| Fig. 1 Photographs of 1 (2.0 wt%) in the presence of Pd2+ (1.0 equiv.) obtained by (a) ultrasonication and (b) heating in acetonitrile. | ||
To understand the role of ultrasound in the gel formation, we observed the UV-vis spectra of the Pd2+–porphyrin metallogel obtained by ultrasonication and the Pd2+–porphyrin sol obtained by heating (Fig. 2). As shown in Fig. 2, the absorption bands of the Pd2+–porphyrin metallogel obtained by ultrasonication appeared at 402 nm, which is the Soret band induced by H-type aggregation (Fig. 2a; see Graphical abstract). In addition, no Q-band was observed in the high wavelength range. This result means that 1 forms a complex with Pd2+ upon ultrasonication. On the other hand, the Soret band of the Pd2+–porphyrin sol obtained by heating was shifted to 490 nm, which is indicative of J-type aggregation (Fig. 2b). In addition, a small amount of H-type aggregation exists upon heating as shown in Fig. 2b. The J-type aggregation obtained by heating was directly converted to H-type aggregation by ultrasonication. These results indicate that the aggregation mode of the Pd2+–porphyrin complex is easily controllable by ultrasonication and heating. In addition, the reversible reaction between H- and J-aggregations was observed by UV-vis spectroscopy (Fig. S1, ESI†).
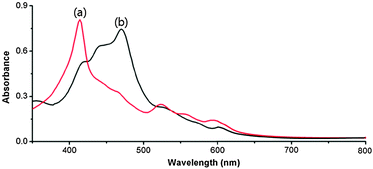 | ||
| Fig. 2 UV-vis spectra of 1 (2.0 wt%) in the presence of Pd2+ (1.0 equiv.) obtained by (a) ultrasonication and (b) heating in acetonitrile. | ||
The absorption intensity of 1 at 402 nm gradually decreased upon the steady addition of aliquots of Pd2+, and it approached saturation at a molar ratio above 1.0 (Fig. S2, ESI†). Furthermore, the Job plot based on the fluorescence changes of the 1–Pd2+ metallogel obtained by ultrasonication indicated 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 binding for 1 with Pd2+ (Fig. S3, ESI†). Using the absorption titration data, we calculated the association constant (K) for Pd2+ coordination with 1 to be 6.3 × 104 M−1.
:1 binding for 1 with Pd2+ (Fig. S3, ESI†). Using the absorption titration data, we calculated the association constant (K) for Pd2+ coordination with 1 to be 6.3 × 104 M−1.
We also examined the effect of temperature on the fluorescence spectra of the H-type aggregation of the Pd2+–porphyrin metallogel obtained by ultrasonication (Fig. S4, ESI†). The fluorescence intensity of the Pd2+–porphyrin metallogel at 652 nm (excitation at 402 nm) increased as the sample was heated until 60 °C, at which point the green color of the metallogel changed to red. These phenomena are attributed to the conversion from the H-type aggregation mode to the J-type aggregation mode. Above 60 °C, the fluorescence intensity decreased again, reflecting the conversion from the J-type aggregation to the monomer species.
For further structural proof for the conversion of the H-type aggregation of the Pd2+–porphyrin metallogel to the J-type aggregation, we observed the PXRD patterns of both the Pd2+–porphyrin metallogel obtained by ultrasonication and the Pd2+–porphyrin complex obtained in the sol state by heating (Fig. S5, ESI†). The main PXRD pattern of the Pd2+–porphyrin metallogel appeared at 15° and 20°–30°, whereas the PXRD pattern of the Pd2+–porphyrin complex obtained in the sol state appeared at 5°, 7°, 12° and 23°, which is quite similar to the pattern of a single crystal of the Pd2+–porphyrin complex. These findings support the view that the aggregation mode of the Pd2+–porphyrin metallogel obtained by ultrasonication is clearly different from that associated with the red color of the Pd2+–porphyrin complex obtained by heating. Below, we will provide a detailed explanation for the single crystal structure of the J-type aggregation of the Pd2+–porphyrin complex obtained in the sol state.
We conducted TEM observations to gain insight into the morphological influence of the Pd2+–porphyrin metallogel obtained by ultrasonication. Surprisingly, as shown in Fig. 3, the superstructures are remarkably different depending upon the gel preparation methods. The Pd2+–porphyrin metallogel prepared by ultrasonication has a sheet-like structure. On the other hand, the Pd2+–porphyrin aggregation after heating showed a cubic structure having a width of 50–80 nm. Furthermore, ultrasonication converted the cubic structure of the Pd2+–porphyrin complex obtained by heating into a sheet-like structure. Thus, the physical properties of the Pd2+–porphyrin superstructures are controllable by ultrasonication and heating.
 | ||
| Fig. 3 TEM images of 1 (2.0 wt%) in the presence of Pd2+ (1.0 equiv.) obtained by (a) ultrasonication and (b) heating. | ||
To investigate the structural differences between the H- and the J-type aggregations, we measured the respective IR spectra of the green colored Pd2+–porphyrin metallogel and the red colored complex (Fig. S6, ESI†). The vibrational intensity of the –COOH group of the Pd2+–porphyrin metallogel is almost the same as that of the –COO−group whereas the vibrational intensity of the –COOH group of the red colored Pd2+–porphyrin complex was half the value of the –COO−group. This suggests that at the molecular level the –COO− species might be induced by J-type aggregation. The vibrational band of the –OH group of the Pd2+–porphyrin metallogel obtained by ultrasonication was of a lower wavenumber than that obtained by heating. This result is attributed to the intermolecular hydrogen-bonding interaction between the carboxylic acid groups of the 1–Pd2+ complex gel obtained by ultrasonication. In addition, the vibrational band of –NO3− of the Pd2+–porphyrin metallogel appeared at 1412 cm−1. This finding is also an indirect evidence for the Pd2+–porphyrin complex.
In addition to the evidence from the IR study, we decided to undertake the preparation and crystal structure of the related compound for further understanding of the driving forces behind the formation of the Pd2+–porphyrin metallogel and also to determine their aggregation modes. We tried to obtain single crystals suitable for X-ray analysis in CH3CN. Unfortunately, the single crystal of 1 in complex with Pd2+ could not be obtained in CH3CN. Thus, the reaction of 1 with K2PdCl4 in aqueous DMF solution was allowed to proceed at 70 °C for 2 days. After cooling to room temperature, we successfully isolated a red complex as single crystals suitable for X-ray analysis. In the complex having the formula [12−·Pd2+], as shown in Fig. 4 and Fig. S7 (ESI†), the palladium(II) center occupying the porphyrin cavity is four-coordinated, typically being bound to the four nitrogen donors adopting a square planar coordination geometry. In packing, notably the complex units prefer to form an offset stacked arrangement, which may be due to the CH⋯π interactions (dashed lines, 2.92–3.15 Å). The observed offset-type packing mode is, at least in part, also stabilized by the H-bonds between carboxylic acids and DMF molecules in the lattice (not shown). Once again, these results indicate that the driving force in the J-type aggregation of 1 with Pd2+ is mainly the CH⋯π interactions between the porphyrin moiety and phenyl protons. We tested the gelation ability of 1–Pd2+ from the single crystal in acetonitrile by ultrasonication. As expected, 1–Pd2+ formed the same gel in acetonitrile. Therefore, we believe that J-aggregation of 1 with Pd2+ in CH3CN provides the same crystal structure as that of a 1–Pd2+ complex obtained from DMF.
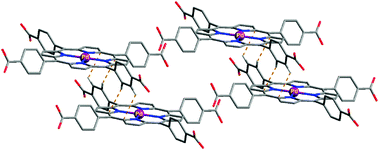 | ||
| Fig. 4 Crystal structure of the palladium(II) complex with 1, showing offset packing arrangement by CH⋯π interactions. The complex was obtained as a red crystal after heating the reaction mixture. | ||
We also carried out dynamic oscillation and steady shear measurements to extend our understanding of the mechanical properties of the Pd2+–porphyrin metallogel. The linear viscoelastic region (LVR) of the Pd2+–porphyrin metallogel, as a function of the amplitude of deformation upon shearing, was determined with a strain amplitude ranging from 0.01% to 100% at 1 rad s−1 (Fig. S8A, ESI†). Both the in-phase storage modulus (G′) and the out-of-phase loss modulus (G′′) remained constant up to ∼1% strain (G′ > G′′), an outcome that defines the uppermost boundary of the LVR. Beyond this level of deformation, a catastrophic disruption of the network occurs as indicated by the steep drop in the values of both moduli and the reversal of the viscoelastic signal (G′′ > G′). Based on these data, we decided to perform subsequent measurements on the gel at 0.1% strain, which lies comfortably within the LVR. Time-dependent oscillation measurements were used to monitor the gelation process of the Pd2+–porphyrin metallogel, which forms gradually upon mixing of porphyrin 1 and Pd2+ by ultrasonication (Fig. S8B, ESI†). The time sweep shows the growth of G′ and G′′ in the initial stage of gelation followed by a slower long-term approach to a final pseudo-equilibrium plateau. At the end of the experiment, the value of G′ was about twice the magnitude of G′′.
In conclusion, we have demonstrated the first example of controlled aggregation models for porphyrin–Pd2+ metallogel formation. We showed that the formation of the metallogel could be facilitated by ultrasonication, which adopts the H-type aggregation mode. On the other hand, heating could induce the J-type aggregation of the porphyrin–Pd2+ complex. The rheological property of the porphyrin–Pd2+ metallogel is similar to those of previously reported typical metallogels. Metallogels may find practical use in various applications, including as scaffold materials in tissue engineering and in catalytic chemistry.
This work was supported by a grant from World Class University (WCU) Program (R32-2008-000-20003-0) and NRF (2011-0005689) supported by Ministry of Education, Science, and Technology, S. Korea.
Notes and references
- Encyclopedia of Supramolecular Chemistry, ed. J. L. Atwood and J. W. Steed, Marcel Dekker, New York, 2004 Search PubMed.
- (a) K. Murata, M. Aoki, T. Suzuki, T. Harada, H. Kawabata, T. Komori, F. Ohseto, K. Ueda and S. Shinkai, J. Am. Chem. Soc., 1994, 116, 6664 CrossRef CAS; (b) M. Ayabe, T. Kishida, N. Fujita, K. Sada and S. Shinkai, Org. Biomol. Chem., 2003, 1, 2744 RSC; (c) S. A. Ahmed, X. Sallenave, F. Fages, G. Gundert, W. M. Müller, U. Müller, F. Vögtle and J.-L. Pozzo, Langmuir, 2002, 18, 7096 CrossRef CAS; (d) L. Frkanec, M. Jokić, J. Makarević, K. Wolsperger and M. Žinić, J. Am. Chem. Soc., 2002, 124, 9716 CrossRef CAS.
- (a) T. Saji, K. Hoshino, Y. Ishii and M. Goto, J. Am. Chem. Soc., 1991, 113, 450 CrossRef CAS; (b) K. Tsuchiya, Y. Orihara, Y. Kondo, N. Yoshino, T. Ohkubo, H. Sakai and M. Abe, J. Am. Chem. Soc., 2004, 126, 12282 CrossRef CAS.
- (a) M. George and R. G. Weiss, Acc. Chem. Res., 2006, 39, 489 CrossRef CAS; (b) N. M. Sangeetha and U. Maitra, Chem. Soc. Rev., 2005, 34, 821 RSC; (c) L. A. Estroff and A. D. Hamilton, Chem. Rev., 2004, 104, 1201 CrossRef CAS; (d) K. J. C. Van Bommel, A. Friggeri and S. Shinkai, Angew. Chem., Int. Ed., 2003, 42, 980 CrossRef.
- T. Naota and H. Koori, J. Am. Chem. Soc., 2005, 127, 9324 CrossRef CAS.
- (a) J. M. J. Paulusse, D. J. M. Van Beek and R. P. Sijbesma, J. Am. Chem. Soc., 2007, 129, 2392 CrossRef CAS; (b) S. Zhang, S. Yang, J. Lan, T. Xue and J. You, J. Am. Chem. Soc., 2009, 131, 1689 CrossRef CAS; (c) W. Wang, J. B. Beck and S. J. Rowan, J. Am. Chem. Soc., 2006, 126, 11663 CrossRef; (d) N. N. Adarsh and P. Dastidar, Cryst. Growth Des., 2011, 11, 328 CrossRef CAS; (e) W. Weng, J. B. Beck, A. M. Jamieson and S. J. Rowan, J. Am. Chem. Soc., 2006, 128, 11663 CrossRef CAS; (f) A. Y.-Y. Tam, K. M.-C. Wong and V. W.-W. Yam, J. Am. Chem. Soc., 2009, 131, 6253 CrossRef CAS; (g) N. Komiya, T. Muraoka, M. Iiida, M. Miyanaga, K. Takahashi and T. Naota, J. Am. Chem. Soc., 2011, 133, 16054–16061 CrossRef CAS.
- N. C. Maiti, S. Mazumdar and N. Periasamy, J. Phys. Chem. B, 1998, 102, 1528 CrossRef CAS.
- R. F. Pasternack, C. Fleming, S. Herring, P. J. Collings, J. dePaula, G. Decastro and E. J. Gibbs, Biophys. J., 2000, 79, 550 CrossRef CAS.
- P. M. R. Paulo and S. M. B. Costa, Photochem. Photobiol. Sci., 2003, 2, 597 RSC.
- P. Kubát, K. Lang, P. Janda and P. Jr Anzenbacher, Langmuir, 2005, 21, 9714 CrossRef CAS.
- Y. Sun, X. Zhang, C. Sun, Z. Wang, J. Shen, D. Wang and T. Li, Chem. Commun., 1996, 2379 RSC.
- K. Ariga, Y. Lvov and T. Kunitake, J. Am. Chem. Soc., 1997, 119, 2224 CrossRef CAS.
- P. G. Van Patten, A. P. Shreve and R. J. Donohoe, J. Phys. Chem. B, 2000, 104, 5986 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Details of experimental section and spectroscopic data. CCDC 843610. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1nj20781d |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2012 |