Water-soluble dendritic polyaspartic porphyrins: potential photosensitizers for photodynamic therapy†
Baoxiang
Gao
*ac,
Yueling
Liu
a,
Huijuan
Yin
b,
Yingxin
Li
*b,
Qianqian
Bai
a and
Licui
Zhang
a
aCollege of Chemistry and Environmental Science, Hebei University, Baoding, 071002, P. R. China. E-mail: bxgao@hbu.edu.cn; Fax: +86 3125079317
bInstitute of Biomedical Engineering, Chinese Academy of Medical Science & Peking Union Medical College, Tianjin, 300192, P. R. China. E-mail: yingxinli@bme.org.cn; Fax: +86 22 87891131
cKey Laboratory of Medicinal Chemistry and Molecular Diagnosis, Ministry of Education, Hebei University, Baoding, 071002, P. R. China
First published on 31st October 2011
Abstract
We prepared these photosensitizers with two-block structures: porphyrin cores for singlet oxygen generation and branched dendritic polyaspartic as hydrophilic shells for inducing water solubility, thus reducing cytotoxicity and giving high singlet oxygen yields.
Photodynamic therapy (PDT) is a new clinical treatment for superficial tumors and age-related muscular degeneration. This technique involves the systemic administration of a photosensitive drug, by which singlet oxygen is generated after light irradiation, to engender oxidative damage to cells.1,2 The PDT strategy utilizes a photosensitizer, working as a light-sensitive drug, to treat the target tissue locally upon the irradiation of light with appropriate wavelengths. One of the key components in effective and efficient PDT is the photosensitizer, also called PDT drugs. The high singlet oxygen quantum yield of the photosensitizers is significantly important for PDT. Most of the conventional photosensitizers have large π-conjugation domains to extend their absorption cross sections and basically have hydrophobic characteristics. Therefore, photosensitizers form aggregates easily, which produce the self-quenching of the excited state in aqueous medium, because of their π–π interaction and hydrophobic characteristics.3,4 A few approaches have been proposed to enhance singlet oxygen quantum yield of the photosensitizers. Quantum dots, gold and silica nanoparticles modified with porphyrin photosensitizer, were developed to undergo energy transfer and to generate singlet oxygen.5–9Polymer micelles encapsulating porphyrin were also prepared to induce singlet oxygen production by the FRET mechanism.10
Recently, Kazunori Kataoka and coworkers reported a dendrimer-based photosensitizer, dendrimer porphyrin (DP), in which the focal porphyrin is surrounded by the third generation of poly(benzyl ether) dendrons.11 Unlike conventional photosensitizers, the DP ensures the efficacy of singlet oxygen production even at an extremely high concentration because the dendritic envelope of DP can prevent aggregation of the central porphyrin.12
Among the requirements for a successful photosensitizer, in addition to singlet oxygen generation capacity, good water solubility and biocompatibility are of paramount importance. Generally, water solubility has been imported by either introducing ionizable hydrophilic groups (carboxylic acid, phosphonic acid, sulfonic acid, and ammonium groups) within the core structure of a fluorescent dye or by grafting the hydrophobic fluorophore to hydrophilic (bio)polymers (carbohydrates, oligonucleotides and polyethylene glycol). Very recently, Akkaya and coworkers reported a series of water-soluble photosensitizers with amino acid modification, and these photosensitizers display significant light-induced cytotoxic effects on the human erythroleukemia cell line.13 Considering that the more biocompatible peptide-based dendrimers have demonstrated promising characteristics as components of vaccines, as well as antiviral and antibacterial agents, we have designed and synthesized dendritic polyaspartic encapsulation of perylene bisimides, which showed good water solubility and biocompatibility.14
In this study, we present the synthesis and property of water-soluble dendritic polyaspartic porphyrins (DPAPs). The chemical structures of DPAPs are shown in Scheme 2. We prepared these DPAPs with two-block structures: porphyrin cores for singlet oxygen generation, and branched dendritic polyaspartic as hydrophilic shells for inducing water solubility and reducing cytotoxicity. These DPAPs are expected to have biocompatibility, water solubility, and high singlet oxygen yields.
Most porphyrin-cored dendrimers have been synthesized through a divergent or a convergent approach. The divergent synthetic approach may often suffer from the disadvantage that the final products contain structural defects. This problem may be avoided by convergent approach. Thus, we chose the convergent approach to dendritic polyaspartic porphyrins. The polyaspartic-dendrons were synthesized according to the procedures reported in the literature.15 The peptide-conjugated benzaldehyde 5, 6 and 7 was obtained by amidation of the carboxybenzaldehyde using equivalent of peptide and the coupling reagents DCC-BtOH in dichloromethane (Scheme 1).
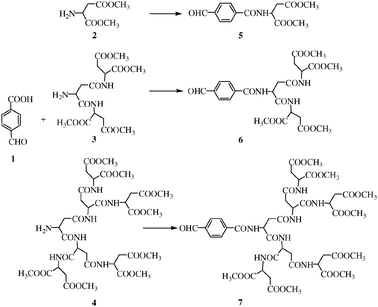 | ||
| Scheme 1 Synthesis of the dendritic polyaspartic substituted benzaldehyde. | ||
The condensation of peptide-conjugated benzaldehyde 5 or 6 with distilled pyrrole using BF3·Et2O as catalyst and dichloromethane as solvent in an aerobic dark environment, following by DDQ oxidation and Et3N neutralization, afforded porphyrins P1-OCH33 and P2-OCH33 with 11% yield (Scheme 2). However, the same reaction conditions did not produce the target compound P3-OCH33 because of the extremely low solubility of the condensation intermediate. By using high boiling point 1,1,2,2-tetrachloroethane, which is usually a good solvent of π-conjugated molecules, the target compound P3-OCH33 was obtained in yields of 13%. Hydrolysis of porphyrins P-OCH33 gave the water-soluble porphyrin compound P-COOH in a nearly quantitative yield. Porphyrin compound P3-COOH could not be precipitated from the aqueous solution even upon strong acidification, and therefore, the solution had to be purified from the excess salt by extensive dialysis. Subsequent lyophilization produced the green powder. Porphyrin compound P3-COOH has in total 28 aspartic units and 32 peripheral carboxylate groups.
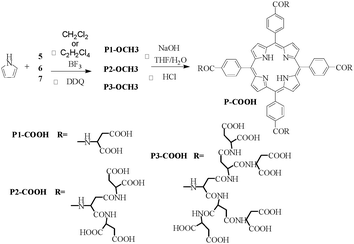 | ||
| Scheme 2 Synthesis of DPAPs. | ||
Optical absorption spectra of porphyrins contain a Soret band (416 nm) and four Q-bands (517, 554, 581, 635 nm) in the UV-vis region (Fig. 1). The spectra of the porphyrin dendrimers resemble those of the cores, although, typical of water-soluble porphyrins, the bands in aqueous solutions are broadened. It appears that the attached dendritic cages seem to have almost no influence on the encapsulated porphyrin absorption bands. The photoluminescence spectra of DPAPs were recorded in 5 × 10−6 mol L−1 aqueous solution (Fig. 2). The porphyrin P1-COOH and P2-COOH have similar fluorescence emission spectra with two distinct peaks at 646 nm and 704 nm. However, the porphyrin P1-COOH shows an additional small peak at 606 nm. Most likely this emission originates exclusively from the mono-protonated porphyrin core.16 Furthermore, we determined the fluorescence quantum yields of the DPAPs in the aqueous solution (5 × 10−6 M). P1-COOH exhibited a low fluorescence quantum yield (8%), which could be attributed to fluorescence quenching by strong aggregation in water.15 With increasing dendron generation, there was a continuous increase in fluorescence quantum yields from 8% for P1-COOH to 11% for P2-COOH and up to 14% for P3-COOH. The bulky dendritic polyaspartic shells may prevent the quenchers from interacting with the porphyrin core.
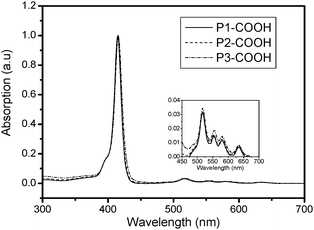 | ||
| Fig. 1 UV-vis absorption spectra of DPAPs in aqueous solutions. | ||
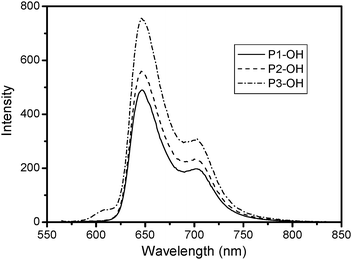 | ||
| Fig. 2 Photoluminescence spectra of DPAPs in aqueous solutions. | ||
Singlet oxygen generation capacities of these DPAPs were studied in water. Relative rates of singlet oxygen generation were evaluated by monitoring the rates of decay of the 1,3-diphenyl-isobenzofuran (the singlet oxygen quencher). Three porphyrin compounds can induce photodegradation of the 1,3-diphenyl-isobenzofuran. Using tetraphenyporphine in benzene as the reference (ΦΔ = 0.63),17 the singlet oxygen yields of compound P1-COOH, P2-COOH and P3-COOH are 0.48, 0.61 and 0.89, respectively. The higher singlet oxygen yield of compound P3-COOH may be ascribed to the branched dendritic polyaspartic groups that somewhat hinder the excited state quench.
The biocompatibility of DPAPs was evaluated for Hela cells using MTT cell-viability assay. Fig. 3 summarizes the viability of Hela cells after being cultured with PEPPs solutions at a concentration of 5.0 × 10−5 for 24 and 72 h. Three porphyrin compounds had very low cytotoxicity (over 90% viability) within 72 h incubation time. It indicates that water-soluble dendritic polyaspartic porphyrins have high biocompatibility. We ascribed the exceptionally low cytotoxicity to dendritic polyaspartic, which protects the dyes from interacting non-specifically with the extracellular proteins and triggering immunogenicity and antigenicity inside the cells.18
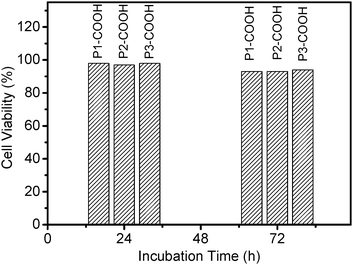 | ||
| Fig. 3 In vitro viability of Hela cells treated with P1-COOH and P2-COOH and P3-COOH solution at 5.0 × 10−5mol L−1 for 24 and 72 h, respectively. | ||
In order to study photodynamic activities of DPAPs, Hela cells were incubated at different dose and then submitted to irradiation (532 nm; 15 mW cm−2) for 20 min, whereas cells not treated by DPAPs and only exposed to the laser were used as controls for cytotoxicity. MTT assay was performed after irradiation, to establish the cytotoxicity of DPAPs. Fig. 4 showed the dose-dependent cells viability for these compounds. It can be seen that the photocytotoxicity depends greatly on the dose. As the concentration increases from 0 μM to 50 μM, the DPAPs showed enhanced photocytotoxicity (cell viability P1-COOH: from 98% to 75%; P2-COOH: from 96% to 25%; P3-COOH: from 94% to 1%). The photocytotoxicity of P3-COOH was much higher than that of P1-COOH and P2-COOH under the same experimental conditions. Furthermore, the photocytotoxicity of DPAPs was in good agreement with the trend observed for the singlet oxygen yields of these compounds.
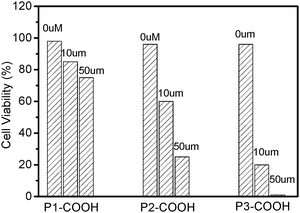 | ||
| Fig. 4 In vitro viability of Hela cells with illumination (532 nm, 15 mW cm−2, 30 J cm−2). | ||
In conclusion, we reported the synthesis and properties of water-soluble dendritic polyaspartic porphyrins. With increasing dendron generation, the singlet oxygen yields of these DPAPs were improved from 48% to 89%. The hydrophilic peptides induced DPAPs with water solubility and biocompatibility. The photodynamic activities of these compounds were further evaluated. These results provide motivation to further explore the anticancer potential of water-soluble dendritic polyaspartic porphyrins.
Experimental
The starting material, 4-carboxybenzaldehyde was purchased from Acros, and used without any further purification. Solvents were purchased from Sinopharm Chemical Reagent Co. Ltd, and used without any further purification. L-Aspartic acid and N-carbobenzyloxy-L-aspartic acids were purchased from Yangzhou Baosheng Bio-Chemical Co. Ltd. N-(3-Dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDCI), 4-dimethylaminopyridine and 1-hydroxybenzotrizole were purchased from Shanghai Medpep Co. Ltd. Compounds 2, 3 and 4 were synthesized according to the reported methods.15 The 1H NMR spectra were recorded at 20 °C on 400 MHz NMR spectrometer (Bruker). Chemical shifts are reported in ppm at room temperature using CDCl3, or DMSO as solvent and tetramethylsilane as internal standard unless indicated otherwise. Abbreviations used for splitting patterns are s = singlet, br = broad, d = dublet, m = multiplet. Mass spectra were carried out using MALDI-TOF/TOF matrix assisted laser desorption ionization mass spectrometry with autoflexIII smartbeam (Bruker Daltonics Inc).Synthesis and characterization of DPAPs
General procedure for synthesis of precursor: dendritic polyaspartic substituted benzaldehyde
0.289 g 4-carboxybenzaldehyde (2 mmol) was dissolved in 60 mL of dichloromethane, 0.575 g 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (3 mmol) was added , and the mixture was stirred at 0 °C for 30 min. 3 mmol polyaspartic-dendrons (3 mmol) and 0.456 g HOBt (3 mmol) were added and the mixture stirred at room temperature for 24 h. Solvent was removed by rotary evaporation. The crude product was purified by column chromatography on silica gel with methylene chloride: ethanol as an eluent to afford dendritic polyaspartic substituted benzaldehyde as a white solid.Compound 5: 66% yield. 1H NMR (CDCl3, 400 MHz): δ 10.09 (s,1 H, CHO), 7.97 (s, 4 H), 7.36 (d, 1 H, CONH), 5.09–5.04 (m, 1 H, CH), 3.81 (s, 3 H, CH3), 3.72 (s, 3 H, OCH3), 3.21–2.96 (m, 2 H, CH2). Anal. Calcd for C14H15NO6: C, 57.34; H, 5.16; N, 4.78. found: C, 57.08; H, 5.27; N, 4.97. m/z [MALDI−TOF]: 294.1 (M+H+).
Compound 6: 51% yield. 1H NMR (CDCl3, 400 MHz): δ 10.09 (s, 1 H, CHO), 8.27–6.89 (m, 3 H, CONH), 7.99–7.76 (m, 4 H, CH), 5.08–4.85 (m, 3 H, CH), 3.76–3.64 (m, 12 H, OCH3), 3.02–2.66 (m, 6 H, CH2). Anal. Calcd for C24H29N3O12: C, 52.27; H, 5.30; N, 7.62. found: C, 52.09; H, 5.41; N, 7.87. m/z [MALDI−TOF]: 552.6 (M+H+).
Compound 7: 51% yield. 1H NMR (DMSO, 400 MHz): δ 10.08 (s, 1 H, CHO), 8.72 (d, J = 7.6 Hz, 1 H, CONH), 8.32–8.99 (m, 2 H, CONH), 8.22–8.16 (m, 4 H, CONH), 8.03–7.97 (m, 4 H, CH), 4.74–4.54 (m, 7 H, CH), 3.60–3.58 (m, 24 H, OCH3), 2.77–2.40 (m, 14 H, CH2). Anal. Calcd for C44H57N7O24: C, 48.49; H, 5.38; N, 9.18. found: C, 48.20; H, 5.50; N, 9.35. m/z [MALDI−TOF]: 1068.8 (M+H+).
General procedure for synthesis of P1-COOH and P2-COOH
To a stirred mixture of dendritic polyaspartic substituted benzaldehyde compound 5 or compound 6 (0.5 mmol) and pyrrole (0.5 mmol) in dichloromethane (200 mL) at room temperature was added boron trifluoride etherate (0.2 mmol). After the mixture was stirred for an additional 2 h, DDQ (1.2 mmol) was added and the reaction mixture was allowed to stir for 30 min at room temperature. The solvent was removed under reduced pressure, and the residue was chromatographed over silica gel using dichloromethane as eluent. The obtained product was redissolved in dichloromethane and precipitated with methanol. Excess of NaOH was added to a solution of compound P1-OCH33 and P2-OCH33 (100 mg) in 10 mL of THF. The mixture was stirred at room temperature for 4 h, and the solvent was evaporated under vacuum at room temperature. Water was added to the remaining solid, and the mixture was stirred at room temperature for additional 2 h. The solution was neutralized by HCl, filtered, dialyzed against distilled water for 4 days, and lyophilized.Compound P1-COOH: 12% yield.1H NMR (DMSO, 400 MHz): δ 12.8 (br, 8 H, COOH), 9.15 (s, 4 H, CONH), 8.88 (s, 8 H, CH), 8.35 (s, 16 H, CH), 4.94–4.92 (m, 4 H, CH), 2.96–2.87 (m, 8 H, CH2), −2.93 (s, 2 H, NH). Anal. Calcd for C64H50N8O20: C, 61.44; H, 4.03; N, 8.96. found: C, 61.23; H, 4.21; N, 9.12. m/z [MALDI−TOF]: 1251.4 (M+H+).
Compound P2-COOH: 11% yield. 1H NMR (DMSO, 400 MHz): δ 12.66 (br, 16 H, COOH), 8.88 (br, 8 H, CH), 8.32 (br, 16 H, CH), 8.88–7.95 (m, 12 H, CONH), 5.00–4.62 (m, 12 H, CH), 2.89–2.73 (m, 24 H, CH2), −2.91 (s, 2 H, NH). Anal. Calcd for C93H90N16O38: C, 54.76; H, 4.45; N, 10.99. found: C, 54.56; H, 4.53; N, 11.10. m/z [MALDI−TOF]: 2040.9 (M+H+).
General procedure for synthesis of P3-COOH
To a stirred mixture of dendritic polyaspartic substituted benzaldehyde compound 7 (0.5 mmol) and pyrrole (0.5 mmol) in 1,1,2,2-tetrachloroethane (200 mL) at room temperature was added boron trifluoride etherate (0.2 mmol). After the mixture was stirred for an additional 2 h, DDQ (1.2 mmol) was added and the reaction mixture was allowed to stir for 30 min at room temperature. The reaction mixture was precipitated with methanol, and washed with chloroform. Excess of NaOH was added to a solution of P3-OCH33 (100 mg) in 10 mL of THF. The mixture was stirred at room temperature for 4 h, and the solvent was evaporated under vacuum at room temperature. Water was added to the remaining solid, and the mixture was stirred at room temperature for additional 2 h. The solution was neutralized by HCl, filtered, dialyzed against distilled water for 4 days, and lyophilized.Compound P3-COOH: 13% yield. 1H NMR (DMSO, 400 MHz): δ 8.88 (s, 8 H, CH), 8.21 (br, 16 H, CH), 9.05–7.97 (m, 28 H, CONH), 4.93–4.45 (m, 28 H, CH), 2.78–2.55 (m, 56 H, CH2), −2.97 (s, 2 H, NH). Anal. Calcd for C160H170N32O92: C, 47.88; H, 4.27; N, 11.17. found: C, 47.69; H, 4.58; N, 11.36. m/z [MALDI−TOF]: 4036.7 (M+Na+).
This work was supported by the Key Project of Chinese Ministry of Education (No. 210015), National Natural Science Foundation of China (No. 20804012), Natural Science Foundation of Hebei Province (No. B2009000169), the Science Research Project of Department of Education of Hebei Province (No. 2007413), and the Natural Science Foundation of Hebei University (No. 2007105).Notes and references
- M. Hoebeke and X. Damoiseau, Photochem. Photobiol. Sci., 2002, 1, 283 CAS.
- X. Damoiseau, F. Tfibel, M. Hoebeke and M. P. Fontaine-Aupart, Photochem. Photobiol., 2002, 76, 480 CrossRef CAS.
- N. Tomioka, D. Takasu, T. Takahashi and T. Aida, Angew. Chem., Int. Ed., 1998, 37, 1531 CrossRef CAS.
- N. Nishiyama, H. R. Stapert, G.-D. Zhang, D. Takasu, D.-L. Jiang, T. Nagano, T. Aida and K. Kataoka, Bioconjugate Chem., 2003, 14, 58 CrossRef CAS.
- G. F. Paciotti, L. Myer, D. Weinreich, D. Goia, N. Pavel, R. E. McLaughlin and L. Tamarkin, Drug Delivery, 2004, 11, 169 CrossRef CAS.
- Y. Cheng, A. C. Samia, J. D. Meyers, I. Panagopoulos, B. Fei and C. Burda, J. Am. Chem. Soc., 2007, 129, 4526 CrossRef.
- S. H. Cheng, C. H. Lee, C. S. Yang, F. G. Tseng, C. Y. Moud and L. W. Lo, J. Mater. Chem., 2009, 19, 1252 RSC.
- S. Shen, A. E. Garcia-Bennett, Z. Liu, Q. Lu, Y. Shi, Y. Yan, C. Yu, W. Liu, Y. Cai, O. Terasaki and D. Zhao, J. Am. Chem. Soc., 2005, 127, 6780 CrossRef CAS.
- S. H. Wu, Y. S. Lin, Y. Hung, Y. H. Chou, Y. H. Hsu, C. Chang and C. Y. Mou, ChemBioChem, 2008, 9, 53 CrossRef CAS.
- W.-D. Jang, N. Nishiyama, G.-D. Zhang, A. Harada, D.-L. Jiang, S. Kawauchi, Y. Morimoto, M. Kikuchi, H. Koyama, T. Aida and K. Kataoka, Angew. Chem., Int. Ed., 2005, 44, 419 CrossRef CAS.
- Y. Li, W. D. Jang, N. Nishiyama, A. Kishimura, S. Kawauchi, Y. Morimoto, S. Miake, T. Yamashita, M. Kikuchi, T. Aida and K. Kataoka, Chem. Mater., 2007, 19, 5557 CrossRef CAS.
- R. Ideta, F. Tasaka, W.-D. Jang, N. Nishiyama, G.-D. Zhang, A. Harada, Y. Yanagi, Y. Tamaki, T. Aida and K. Kataoka, Nano Lett., 2005, 5, 2426 CrossRef CAS.
- F. Yukruk, A. L. Dogan, H. Canpinar, D. Guc and E. U. Akkaya, Org. Lett., 2005, 7, 2885 CrossRef CAS.
- B. X. Gao, H. X. Li, H. M. Liu, L. C. Zhang, Q. Q. Bai and X. W. Ba, Chem. Commun., 2011, 47, 3894 RSC.
- B. X. Gao, H. X. Li, D. F. Xia, S. F. Sun and X. W. Ba Beilstein, J. Org. Chem., 2011, 7, 198 CAS.
- S. A. Vinogradov and D. F. Wilson, Chem.–Eur. J., 2000, 6, 2456 CrossRef CAS.
- M. C. DeRosa and R. J. Crutchley, Coord. Chem. Rev., 2002, 233–234, 351 CrossRef CAS.
- J. M. Harris and R. B. Chess, Nat. Rev. Drug Discovery, 2003, 2, 214 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: See DOI: 10.1039/c1nj20733d |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2012 |