Graphene oxide microspheres prepared by a simple, one-step ultrasonication method†
Matias
Sametband
,
Ulyana
Shimanovich
and
Aharon
Gedanken
*
Department of Chemistry, Kanabar Laboratory for Nanomaterials, Institute of Nanotechnology and Advanced Materials, Bar Ilan University, Ramat Gan, Israel 52900. E-mail: gedanken@mail.biu.ac.il; Fax: +972 37384053; Tel: +972 35318315
First published on 11th October 2011
Abstract
We demonstrate herein a simple, one-step method for preparing stabilized microspheres of graphene oxide (GO), by applying ultra-sonic power to a biphasic system. The microsphere's size was affected by the pH of the aqueous solution, ranging from a few mm to μm. Further characterization indicated that the microsphere's inner content is composed mainly of organic solvents, though water and GO molecules may be also present at the microsphere's core. The microspheres were stable for several months without a significant conformation change. We predict that the stability arises from hydrophobic and hydrophilic interactions between the GO sheets and the solvents. Changing the organic solvent resulted in changes in the microsphere's morphology.
Graphene and graphene oxide (GO) have become promising materials for biology,1 optical,2 nanocomposites,3 and electronic applications.4 GO and graphene are mainly produced by chemical oxidation and exfoliation of graphite flakes, followed by GO post-reduction to graphene (rGO).5 The oxidation process results in a water-soluble GO suspension, due to the presence of oxygen-containing functional groups on the GO surface. On the other hand, hydrophobic regions are present at the GO's basal plane originating from graphitic sp2 clusters.6 Micro- and nanospheres were previously synthesized through ultrasonic emulsification. Different proteinaceous microspheres were prepared from various proteins, such as bovine serum albumin (BSA), human serum albumin (HSA), hemoglobin and also mixed proteins.7 We recently reported on the preparation of starch, chitosan, DNA and RNA micro- and nanopheres.8 Graphene and GO microspheres were previously prepared by several methods: hollow graphene oxide (GO) microspheres were prepared using a water-in-oil system, through heating and removal of the water phase.9 In addition, superhydrophobic microspheres of polymer–graphene composites were formed by gelation.10
We report herein on a new, simple method for the preparation of liquid-filled GO microspheres using ultra-sonication. GO was selected due to its amphiphilic nature, which we predicted should allow the preparation of stable, liquid-filled microspheres.
The graphene oxide (GO) synthesis resulted in the formation of a water soluble, single layer GO (see the ESI†, Fig. S1). UV-vis absorbance indicated that graphite was oxidized to GO, with a typical absorption peak at ∼237 nm. XPS and IR measurements indicated that oxygen derivatives were present, confirming the graphite oxidation.11 XRD diffraction pattern peaks for pristine graphite and GO (26.6° and 10.5°, respectively) corresponded to previously reported diffractions.12 Raman spectra showed typical GO G peak (∼1590 cm−1) and D peak (∼1350 cm−1). TEM and AFM images pointed out that the exfoliated GO was single layer GO sheets, with a typical thickness of ∼1.1 nm.13
GO microspheres were prepared through ultrasonic emulsification, using a biphasic system of GO in double-distilled water (DDW) and dodecane. Fig. 1 represents an example of the emulsification process using ultrasound: three phases were formed, in which the microspheres were present between the lower aqueous phase and the organic upper phase.
 | ||
| Fig. 1 GO microspheres prepared in three different aqueous pHs: (a) acidic, (b) neutral and (c) alkaline. As the pH increased, smaller microspheres were formed, and more GO remained soluble in the aqueous phase. | ||
pH variations of the aqueous phase (2, 6.5, 12) resulted in different microsphere sizes. While at acidic and neutral pH the microspheres diameter was between hundreds of μm to a few mm (Fig. 1a and b) and the microspheres were visible to the naked eye, at alkaline pH the microspheres were significantly smaller (Fig. 1c). Their size was determined by light microscopy, and the images indicated a poly-dispersed size distribution, with an average diameter of 7 ± 3 μm (Fig. 2a). Scanning electron microscope (SEM) imaging illustrated that the microspheres diameter coincides with the light microscope images, albeit slightly smaller (Fig. 2b). The outline of the microspheres in this case was slightly elliptic, probably due to the interaction between the microspheres and the silicon substrate. The microspheres were stable for several months, with no significant changes in their size (see the ESI†, Fig. S2), although they collapsed after drying.
 | ||
| Fig. 2 (a) Light microscope image of GO microspheres prepared at alkaline pH. The microspheres size was poly-dispersed, with an average diameter of 7 ± 3 μm (magnification ×40). (b) SEM images of GO microspheres on a Si/SiO2 substrate, coated with a thin layer of gold. | ||
Interestingly, as the pH was increased, more soluble GO was present in the lower aqueous phase after the sonication process. From absorbance measurements of the aqueous solution before and after sonication, we calculated the percent of GO left in the aqueous solution after sonication. At acidic and neutral pHs, the GO concentration in the solution decreased drastically compared to the initial concentration (see the ESI†, Fig. S3, 2% and 19%, respectively, initial concentration regarded as 100%). At alkaline pH the reduction was minor, and 91% of GO was still present in the aqueous phase after sonication. GO is regarded as an amphiphilic molecule, having a high surface activity, which is pH dependent.14,15 As the pH of the solution increases, the GO becomes more hydrophilic. It is suggested that due to the higher water solubility, less GO was involved in the microsphere creation in the alkaline solution, and part of the GO is found in the bottom aqueous phase. In contrast, at lower pH values, the GO was more hydrophobic and tended to better interact with the hydrophobic phase and be less soluble in the aqueous phase, resulting in bigger microspheres.
Due to the amphiphilic nature of GO, we were interested in understanding if the microspheres were water-in-oil (w/o) or oil-in-water (o/w) emulsions. For this purpose, Oil Red O (ORO) dye was dissolved in the organic phase (insoluble in water), followed by ultra-sonication at alkaline pH. The dodecane solution absorbance was measured before and after the sonication reaction (λmax ≈ 518 nm). ORO absorbance was reduced by ∼25% as compared to the absorbance prior to the reaction (see the ESI†, Fig. S4). The absorbance reduction suggested that the content in the microsphere's core was composed of dodecane, suggesting that o/w emulsions were formed, although the presence of water molecules inside the microspheres could not be excluded. We continued our study by using the solvatochromic fluorescent dye Nile Red (NR). NR's excitation and emission spectra are affected by the environment polarity in which the NR molecules are dissolved.16 As the solvent's dielectric constant increases, the excitation and emission maxima are shifted towards longer wavelengths. In our case, if the microsphere core contains dodecane, the dye should emit green fluorescence. On the other hand, if a polar environment is present at the core, the NR dye should emit red fluorescence. The fluorescence images (Fig. 3) clearly indicated a strong green fluorescence emitted from the microsphere's core, suggesting a non-polar environment due to the presence of dodacane (Fig. 3c). By changing the excitation wavelength, we noticed that red fluorescence was also emitted from the microsphere's core (Fig. 3d). Fluorescence profile measurements revealed that the intensity of the red fluorescence was stronger close to the microsphere's edges compared to the core, although red fluorescence was also emitted from inside the core (Fig. 3e). This was in contrast to the green fluorescence intensity, which was stronger at the center of the core (Fig. 3g). The red fluorescence indicated that the polarity inside the microsphere increased towards the edges. As mentioned before, the GO is an amphiphilic molecule, attracting water molecules in its vicinity, and so we expect water molecules, if present inside the spheres, to be located closer to the GO shell. We predict that water molecules may also be present to some extent at the microsphere's core, resulting in a local polar environment. GO's oxygen-containing groups may also be responsible for the polar environment at the microsphere shell. It should be noticed that water tends to quench NR fluorescence to some extent,16 and so we predict that the red fluorescence at the edge is mainly due to the polar environment caused mostly by the GO sheets.
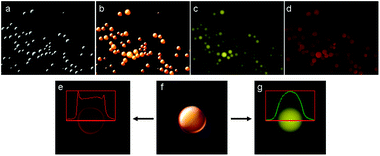 | ||
| Fig. 3 Fluorescence images of GO microspheres synthesized in the presence of nile red (magnification ×40). (a) White light mode, (b) combined image of white light and fluorescence images, (c) fluorescence image using a Yellow Fluorescent Protein filter (YFP, λex = 500 ± 25 nm, λem = 535 ± 30 nm), (d) fluorescence image using a Texas Red filter (TR, λex = 560 ± 25 nm, λem = 615 ± 20 nm), (e) fluorescence profile measurement using the TR filter, showing an increase in red fluorescence close to the microsphere's edge, (f) combined image of white light and fluorescence images, (g) fluorescence profile measurement using the YFP filter, indicating an increase in fluorescence at the microsphere's core. | ||
We predict from these results that the microsphere's core is composed of a mixture of mostly dodecane, water and possibly GO molecules. The presence of water molecules at the microspheres core may hint on the size difference between the different pH conditions. Water present at the core and edges interacts with the negatively charged GO sheets, stabilizing the microsphere at alkaline pH, resulting in smaller microsphere diameters.
We performed the sonication process with two additional initial GO concentrations: 1 mg and 0.3 mg GO in 30 ml DDW, all performed at alkaline pH (12). Changing the initial GO concentration had an effect neither on the microsphere's size, nor on the volume of the microsphere's phase (see the ESI†, Fig. S5). This may result from microsphere saturation during the sonochemical reaction, in which the amount of microspheres created from a specific volume ratio of DDW![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :dodecane is constant at a certain pH. The microsphere's diameter seems to be affected primarily from the solution's pH and not from the initial GO concentration, as long as the GO is in excess.
:dodecane is constant at a certain pH. The microsphere's diameter seems to be affected primarily from the solution's pH and not from the initial GO concentration, as long as the GO is in excess.
During ultra-sonication, micron-size gas bubbles or non-aqueous droplets are formed in the aqueous solution,17 around which the microspheres are formed. This phenomenon is a result of the interaction between the sound waves traveling through the liquid, and gas molecules present in the solvent. The gas type and content present in the solvent affect the bubble formation and collapse process.18 Monoatomic gases, such as argon, provide a larger sonochemical effect compared to biatomic gases, which should affect the microsphere formation. We performed the sonication emulsification in the presence of argon, at pH 12, and to our surprise no affect was noticed (see the ESI†, Fig. S6). The microsphere's diameter was similar to the spheres prepared under ambient air conditions.
The duration of the sonication process was varied from 3 min to 10 and 30 min (pH 12). No changes in the microsphere morphology were observed in this time range. There was a slight difference in the GO concentration present in the aqueous phase after sonication for 30 min, which was reduced to 82% compared to the initial concentration (see the ESI†, Fig. S3). This indicated that prolonging the duration of the sonication time results in a slight increase in microsphere creation.
The amphiphilic nature of the GO may allow the formation of micelles in a biphasic system without sonication. We were interested in testing whether small GO microspheres may form by vigorous stirring, with no sonication applied. The results indicated that at acidic and neutral pH large, millimetre size spheres were formed, which were drastically bigger as compared to the microspheres prepared by the sonochemical method (see the ESI†, Fig. S7). At alkaline pH, almost no spheres were created, probably due to the high GO solubility in the aqueous phase. In all cases most of the GO was found in the aqueous phase after sonication, and at acidic pH it precipitated with time. This indicated that the sonication process has major importance in the formation of micron-size GO spheres.
Microspheres were prepared using toluene and dichloromethane (DCM) as the organic phases, to test whether the nature of the organic phase affects the microsphere's morphology. Toluene was expected to create π–π interactions with the GO sheets, while DCM has a higher polarity and density than dodecane. When toluene was used, similar results to the dodecane reaction were achieved at acidic and neutral pH (see the ESI†, Fig. S8). On the other hand, when the sonication process was performed under alkaline conditions, large spheres were formed with diameters of a few mm (Fig. 4a), in contrast to the μm-size microspheres created with dodecane. These large spheres were stable for months. We predict that due to the high hydrophilic nature of GO at alkaline pH, repulsion occurs between the toluene molecules and the GO sheets, resulting in bigger spheres. When DCM was used, no spheres were produced at acidic or neutral pH. Only in an alkaline environment large spheres were formed, although smaller than in the toluene case (Fig. 4b). The DCM spheres were stable for 2 weeks, indicating that the solvent–GO interaction is important for sphere stability.
 | ||
| Fig. 4 GO microspheres prepared with different organic solvents: toluene (a) and dichloromethane (b), pH 12. | ||
It was previously reported that microspheres stability depended on acquiring a more stable conformation (starch microspheres) or the creation of disulfide linkages when proteins were used.8 These linkages were formed by radical generation during the sonication process. In the case of GO, we believe that the main interactions are hydrophilic and hydrophobic interactions between the GO sheets, and between the GO and the hydrophobic and hydrophilic environments at the microsphere's core, which are pH dependent. This conclusion coincides with previous reports on the importance of hydrophobic interactions when streptavidin and poly-glutamic acid microspheres were prepared.19 In addition, the creation of inter-linkages between the GO sheets due to radical generation may also stabilize the GO microspheres.
In conclusion, we have demonstrated a new procedure for preparing liquid-filled GO microspheres using a simple, biphasic sonication method. pH has a crucial effect on the morphology and diameter of the microspheres created. We showed that the initial GO concentration is less important than the solution's pH. By using two dyes we were able to show that the microsphere's core has mostly a hydrophobic environment. A polar environment was also measured at the microsphere's core, which increased close to the microsphere edge. This can be attributed to the presence of GO sheets and water molecules. Different organic solvents resulted in different behavior during the ultra-sound reaction, resulting in different microsphere sizes. A full study is currently being undertaken for the purpose of understanding the mechanism behind GO microsphere formation during the ultra-sonication process.
Experimental
Preparation of GO microspheres
3 mg GO was added to 30 ml double distilled water (DDW) and exfoliated by ultra-sonication. After the GO was exfoliated, the aqueous suspension was either untreated (pH = 6.5), acidified (pH = 2) with 1 M aqueous HCl, or brought under alkaline conditions (pH = 12) with 1 M aqueous NaOH. These three experiments were continued in parallel by adding 20 ml of 99% anhydrous dodecane, resulting in two distinct phases. The sonicator's horn was immersed into the solution, and the horn's tip was placed at the interface between the two phases. The solution was cooled with an ice bath (temperature kept below 30 °C), and the sonication power was applied for 3 min with 40% efficiency. The solution was stored at rt for three days for full phase separation before further characterization. Oil Red O dye (ORO) containing microspheres were prepared by adding 0.01% (w/v) ORO to the dodecane phase, and performing the sonication as reported above (alkaline conditions). Nile Red (NR) containing microspheres were prepared by dissolving 0.5 mg NR in 20 ml dodecane, stirred for 30 min, and added to the exfoliated GO solution (pH = 12). The sonication was performed in a similar manner as reported above. The microspheres were washed several times with dodecane to remove any excess of NR, followed by dilution with DDW prior to visualization.Characterization
GO was characterized through several methods: the atomic force microscope (AFM) measurements and imaging were carried out using a Nanoscope V Multimode scanning probe microscope (Digital Instruments, Santa Barbara, CA), in which a drop of GO suspension was cast on a Si/SiO2 substrate (cleaned with piranha solution); transmission electron microscope (TEM) images were acquired using a Philips CM-120 operating at 120 kV; X-ray diffraction (XRD) measurements were performed with a Bruker D8 diffractometer (Karlsruhe, Germany); X-ray photoelectron spectroscopy (XPS) measurements were done with an Axis HS with a monochromatic Al K source (Kratos Analytical); Raman spectra were obtained with a JY Horiba Olympus Bx41 spectrometer (Longjumeau, France); FT-IR spectra was acquired by a Jasco Inc. FT-IR 4200; and UV-vis absorption spectra was acquired by a Cary 100 Scan UV spectrophotometer. Microsphere size determination was conducted by light microscopy, employing an Axio Imager Z1, Zeiss optical microscope. Fluorescence imaging was performed at two different excitation and emission ranges, using a Yellow Fluorescent Protein filter (YFP, λex = 500 ± 25 nm, λem = 535 ± 30 nm) and a Texas Red filter (TR, λex = 560 ± 25 nm, λem = 615 ± 20 nm). Scanning electron microscopy (SEM) was performed with a JSM-840, JEOL at an accelerating voltage of 3 kV, the microsphere samples were fixated with glutaraldehyde on a Si/SiO2 surface, dried and coated with a thin gold layer.Notes and references
- X. Sun, Z. Liu, K. Welsher, J. T. Robinson, A. Goodwin, S. Zaric and H. Dai, Nano Res., 2008, 1, 203–212 CrossRef CAS.
- K. P. Loh, Q. Bao, G. Eda and M. Chhowalla, Nat. Chem., 2010, 2, 1015–1024 CrossRef CAS.
- (a) Y. Cao, Z. Lai, J. Feng and P. Wu, J. Mater. Chem., 2011, 21, 9271–9278 RSC; (b) D. Cai and M. Song, J. Mater. Chem., 2010, 20, 7906–7915 RSC.
- (a) L. Kavan, J. H. Yum and M. Gratzel, ACS Nano, 2011, 5, 165–172 CrossRef CAS; (b) Y. Ji, S. Lee, B. Cho, S. Song and T. Lee, ACS Nano, 2011, 5, 5995–6000 CrossRef CAS.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- N. Lu, D. Yin, Z. Li and J. Yang, J. Phys. Chem. C, 2011, 115, 11991–11995 CAS.
- (a) K. S. Suslick and M. W. Grinstaff, Abstr. Pap. Am. Chem. Soc., 1994, 207 Search PubMed , 57-INOR; (b) K. S. Suslick and M. W. Grinstaff, Abstr. Pap. Am. Chem. Soc., 1991, 201 Search PubMed , 197-POLY; (c) M. Wong and K. S. Suslick, Abstr. Pap. Am. Chem. Soc., 1995, 210 Search PubMed , 587-INOR; (d) U. Angel (Shimanovich), D. Matas, S. Michaeli, A. Cavaco-Paulo and A. Gedanken, Chem.–Eur. J., 2010, 16, 2108–2114 CrossRef.
- (a) O. Grinberg and A. Gedanken, Macromol. Chem. Phys., 2010, 211, 924–931 CAS; (b) N. Skirtenko, T. Tzanov, A. Gedanken and S. Rahimipour, Chem.–Eur. J., 2010, 16, 562–567 CrossRef CAS; (c) U. Shimanovich, V. Volkov, D. Eliaz, A. Aizer, S. Michaeli and A. Gedanken, Small, 2011, 7, 1068–1074 CrossRef CAS; (d) U. Shimanovich, D. Eliaz, A. Aizer, I. Vayman, S. Michaeli, Y. Shav-Tal and A. Gedanken, ChemBioChem, 2011, 12, 1678–1681 CrossRef CAS.
- P. Guo, H. Song and X. Chen, J. Mater. Chem., 2010, 20, 4867–4874 RSC.
- L. Zhang, D. A. Zha, T. Du, S. Mei, Z. Shi and Z. Jin, Langmuir, 2011, 27, 8943–8949 CrossRef CAS.
- J. I. Paredes, S. Villar-Rodil, A. Martinez-Alonso and J. M. D. Tascon, Langmuir, 2008, 24, 10560–10564 CrossRef CAS.
- R. Y. N. Gengler, A. Veligura, A. Enotiadis, E. K. Diamanti, D. Gournis, J. Csaba, B. J. van Wees and P. Rudolf, Small, 2010, 6, 35–39 CrossRef CAS.
- J. Zhao, S. Pei, W. Ren, L. Gao and H. M. Cheng, ACS Nano, 2010, 4, 5245–5252 CrossRef CAS.
- F. Kim, L. J. Cote and J. Huang, Adv. Mater., 2010, 22, 1954–1958 CrossRef CAS.
- J. Kim, L. J. Cote, F. Kim, W. Yuan, K. R. Shull and J. Huang, J. Am. Chem. Soc., 2010, 132, 8180–8186 CrossRef CAS.
- P. Greenspan and S. D. Fowler, J. Lipid Res., 1985, 26, 781–789 CAS.
- A. Gedanken, Chem.–Eur. J., 2008, 14, 3840–3853 CrossRef CAS.
- Applied Sonochemistry: The Uses of Power Ultrasound in Chemistry and Processing, ed. T. J. Mason and J. P. Lorimer, Wiley-VCH Verlag GmbH, Weinheim, Germany, 2002 Search PubMed.
- (a) S. Avivi (Levi) and A. Gedanken, Ultrason. Sonochem., 2005, 12, 405–409 CrossRef CAS; (b) E. M. Dibbern, F. J. J. Toublan and K. S. Suslick, J. Am. Chem. Soc., 2006, 128, 6540–6541 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: GO synthesis and exfoliation, characterization, microsphere stability, ORO, sonication duration, and different initial GO concentration results. See DOI: 10.1039/c1nj20738e |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2012 |