Catalytic performance of lanthanide molecular solids containing well-modified metal–organic octahedra†
Xiao
Wu
,
Zhihua
Lin
,
Cheng
He
* and
Chunying
Duan
State Key Laboratory of Fine Chemicals, Dalian University of Technology, Dalian 1160, 24, China. E-mail: hecheng@dlut.edu.cn
First published on 15th November 2011
Abstract
Channel-like and cage-like porous molecular materials Tb–PT1 and Sm–PT1 were constructed through packing and stacking of the amide-containing lanthanide-based octahedra [Tb6(H3L)4 (NO3)9-3H+]6+ and [Sm6(H3L)4(NO3)12]6+, respectively, where H3L is N′,N′,N′′-tris(pyridin-2-ylmethylene)benzene-1,3,5-tricarbohydrazide. Catalytic experiments on cyanosilylation reactions exhibited that the loading of only 1 mol% of Tb–PT1 leads to >90% conversion of the products with all the three nitrobenzaldehydes (NBA), but less than 10% conversion in the case of large size 3-formyl-1-phenylene-3 and 5-di-tert-butylbenzoate (BA) substrates. Adsorption of the substrates in the catalyst demonstrated a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mole ratio (per octahedron) of these nitrobenzaldehydes, but no adsorption of BA could be observed. Luminescent detection of the suspension of Tb–PT1 in CH2Cl2 solution exhibited that the presence of nitrobenzaldehydes quenched the emissions efficiently, whereas the addition of BA did not cause any significant spectra variations. All these results revealed that the catalytic performance of Tb–PT1 was size-selective and the interactions corresponding to the Tb3+ sites were dominant in activating the aldehydes. The catalytic experiments on aldol reactions between cyclohexanone and these aldehydes exhibited that the smallest 4-NBA substrate has the largest conversion (80%) over the others. The presence of cyclohexanone led to absorption spectral changes and luminescence enhancements of the catalyst. These results suggested that interactions between cyclohexanone and the amide groups were dominant in activating the cyclohexanone and the aldol reactions possibly occurred within the cavities of the octahedron. Evaluation of catalytic performance of Sm–PT1 in which those channels in Tb–PT1 were blocked upon cyanosilylation and aldol reactions as well as the adsorption experiments were also carried out for a comparison.
:1 mole ratio (per octahedron) of these nitrobenzaldehydes, but no adsorption of BA could be observed. Luminescent detection of the suspension of Tb–PT1 in CH2Cl2 solution exhibited that the presence of nitrobenzaldehydes quenched the emissions efficiently, whereas the addition of BA did not cause any significant spectra variations. All these results revealed that the catalytic performance of Tb–PT1 was size-selective and the interactions corresponding to the Tb3+ sites were dominant in activating the aldehydes. The catalytic experiments on aldol reactions between cyclohexanone and these aldehydes exhibited that the smallest 4-NBA substrate has the largest conversion (80%) over the others. The presence of cyclohexanone led to absorption spectral changes and luminescence enhancements of the catalyst. These results suggested that interactions between cyclohexanone and the amide groups were dominant in activating the cyclohexanone and the aldol reactions possibly occurred within the cavities of the octahedron. Evaluation of catalytic performance of Sm–PT1 in which those channels in Tb–PT1 were blocked upon cyanosilylation and aldol reactions as well as the adsorption experiments were also carried out for a comparison.
Introduction
Metal–organic polyhedra (MOPs), discrete molecular architectures constructed through the coordination of metal ions and organic linkers, have attracted considerable attention due to their high symmetry, stability and rich chemical/physical properties potential for a variety of applications.1,2 These molecules are synthesized by using modular and high yield metal directed self-assembly methods, so that the geometric and electronic characteristics embedded within the individual components have collectively allowed the construction of the supramolecular entities in a controllable way.3,4 These structures discussed exhibit well-defined cavities with gated pores providing specific inner environments for selective uptaking and binding of guest molecules. Many of them exhibit reactivity and specificity reminiscent of natural systems, and some of them have functions that exceed the natural systems which provided the inspiration for initially making them.5,6 For example, the special micro-environment in the cage-compounds of Raymond et al.7 increases dramatically the acid-catalyzed hydrolysis reaction of orthoesters even in a basic reaction medium. Fujita applied the typical M6L4 cage-compounds in Diels–Alder reactions to obtain unique products through the cage-directed selectivity effect.8 These recent breakthroughs also demonstrate that the implementation of supramolecular strategies in traditional homogeneous catalysis approaches serves as a powerful tool to solve problems in the field of homogeneous catalysis.9The immobilization of homogeneous catalysts on solid supports represents an established field that is on the verge of being applied in industry,10 and the emerging young field of microporous metal–organic frameworks (MOFs) has attracted wide attention for their superior functional properties and applications in heterogeneous catalysis.11 The use of metal–organic macrocycles as molecular building blocks for the creation of extesively porous materials has implications for a whole generation of heterogeneous catalysts. Since the molecules defining the cavities conceivably align and stack in the solid state to create semi-infinite channel structures, these materials would be expected to typically display the combined advantages of homogeneous and heterogeneous catalyses,12 such as high activity and selectivity on the one hand, and easy separation and efficient recycling, on the other, that result in an overall lower cost compared to others.13 The ready tenability of such an approach is also expected leading to unique and useful heterogeneous catalysis, since the rigid and shape-persistent metal–organic macrocycles are versatile building blocks for supramolecular design and are possibly suited for the hierarchical assembly of porous materials with utilizable functional groups.
Through carefully incorporating amide groups as guest-accessible functional organic sites within the well-defined metal–organic architectures, researchers, including us, have reported a powerful approach to assemble metal-tunable Werner-type cages composed of transition metal and lanthanide ions for size- or shape-selective dynamic molecular sensing.14 In several recent discoveries, amide-containing metal–organic polyhedra (MOPs)13c,15 and MOFs16 were found to be capable of prompting chemical reactions efficiently. Since the coordinated unsaturated lanthanide ions are also able to function as active catalysis sites for acid promoting reactions,17 we envisioned that the assembled metal–organic polyhedra containing both potential unsaturated lanthanide ions and amide groups should exhibit efficient base-type and acid-type catalytic performances at the same time. To realize the above-mentioned postulation, herein we investigate the preparation of porous materials through the packing and stacking of the amide-containing lanthanide-based octahedra Tb–PT1 and Sm–PT1 in the crystalline solid14 and validate the catalytic properties in cyanosilylation reactions and aldol reactions, respectively.
Experimental
Instruments and reagents
All chemicals were of reagent grade quality, obtained from commercial sources and used without further purification. The elemental analyses of C, H and N were performed on a Vario EL III elemental analyzer. 1H NMR spectra were measured on a Varian INOVA 400 M spectrometer. The powder XRD diffractograms were obtained on a RigukuD/Max-2400 X-ray Diffractometer with a Cu sealed tube (λ = 1.54178 Å).Preparation of Tb–PT1
The synthetic procedure of complex Tb–PT1 has been reported in our previous work.14 Anal. calc. for Tb6(C108H81N36O12)(NO3)15(H2O), H 2.10, C 32.62, N 17.96%; found: H 2.70, C 32.12, N 17.43%. 1H NMR (DMSO-d6, ppm): 12.48 (s, 9H, H2), 8.63 (br, 36H, H3, H7, H1), 8.04 (br, 24H, H4, H5), 7.45 (br, 12H, H6). IR (KBr plate): 3421 (br, υN–H and υO–H), 1636 (m, υC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and υCN), 1560, 1517, 1476 (w, υAr–CC– and υAr–CN–), 1384 (s, υNO3), 1300 (m, υC–N).
O and υCN), 1560, 1517, 1476 (w, υAr–CC– and υAr–CN–), 1384 (s, υNO3), 1300 (m, υC–N).
Preparation of Sm–PT1
A solution of Sm(NO3)3·6H2O (0.033 g, 0.075 mmol) in methanol (6 mL) was layered onto a solution of PT1 ligand (0.026 g, 0.05 mmol) in CH3OH/CHCl3 (v:v = 1:4, 6 mL). The solution was left for one week at room temperature to give X-ray quality yellow block crystals. Yield: about 62% based on the crystals that have been collected and then dried in vacuum. Anal. calc. for Sm6(C27H21N9O3)4(NO3)18, H 2.07, C 31.67, N 18.46%; found: H 2.55, C 30.94, N 17.80%. 1H NMR (DMSO-d6, ppm): 12.49 (s, 12H, H2), 8.65 (m, 36H, H3, H7, H1), 8.05 (d, 12H, H4, J = 7.2 Hz), 7.93 (t, 12H, H5, J = 15.3 Hz), 7.47 (t, 12H, H6, J = 11.7 Hz). IR (KBr plate): 3398 (br, υN–H and υO–H), 1636 (m, υCO and υCN), 1560, 1516, 1474, 1439 (w, υAr–CC– and υAr–CN–), 1384 (s, υNO3), 1297 (m, υC–N).
General procedure for the aldol reaction
Compound Tb–PT1 or Sm–PT1 (10 μmol) was added to a suspension of aldehyde (0.5 mmol), cyclohexanone (0.52 mL, 5 mmol), water (0.5 mL) and CH3OH (0.5 mL) at 37 °C. The reaction mixture was stirred for 5 d and EtOAc (5 mL) was added. The filtrate was dried (Na2SO4) and concentrated in a vacuum. The conversion and diastereomer were determined by 1H NMR analysis of the crude aldol products.General procedure for the cyanosilylation of aldehyde substrates
Compound Tb–PT1 or Sm–PT1 (5 μmol) was added to a suspension of aldehyde (0.5 mmol), Me3SiCN (0.594 g, 0.6 mmol) and dry dichloromethane (0.5 mL) at 20 °C. The reaction mixture was stirred for 1.5 h and dry dichloromethane (5 mL) was added. The filtrate was concentrated in a vacuum. The conversion was determined by 1H NMR analysis of the crude products.Crystallography
X-Ray intensity data were measured at 180(2) K on a Bruker SMART APEX CCD-based diffractometer (Mo-Kα radiation, λ = 0.71073 Å) using the SMART and SAINT programs. The structures were solved by direct methods and refined on F2 by full-matrix least-squares methods with SHELXTL version 5.1. Crystal data of Sm–PT1: Sm6(C27H21N9O3)4(NO3)18·5CHCl3·CH3OH·9H2O (C114H111Cl15 N54O76Sm6), M = 4887.42, hexagonal, space group R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) , yellow block, a = 28.803(4), c = 44.831(9) Å, V = 32211(9) Å3, Z = 6, Dc = 1.497 g cm−3, μ(Mo-Kα) = 1.890 mm−1, T = 180(2) K. 11317 unique reflections [Rint = 0.0722]. Final R1 [with I > 2σ(I)] = 0.0791, wR2 (all data) = 0.2605. Non-hydrogen atoms were refined anisotropically. CCDC number: 675067. Except the solvent molecules, hydrogen atoms were fixed geometrically at calculated distances and allowed to ride on the parent non-hydrogen atoms with the isotropic displacement being fixed at 1.2 times of the atoms they attached to. Several solvent CHCl3 molecules were refined with the C–Cl bond distance being fixed at 1.57 Å and Cl⋯Cl separation of 2.50 Å, respectively. One of the uncoordinated nitrate anions was refined with the N–O bond distance being fixed at 1.25 Å and the separation between three oxygen atoms was fixed to be the same, respectively.
, yellow block, a = 28.803(4), c = 44.831(9) Å, V = 32211(9) Å3, Z = 6, Dc = 1.497 g cm−3, μ(Mo-Kα) = 1.890 mm−1, T = 180(2) K. 11317 unique reflections [Rint = 0.0722]. Final R1 [with I > 2σ(I)] = 0.0791, wR2 (all data) = 0.2605. Non-hydrogen atoms were refined anisotropically. CCDC number: 675067. Except the solvent molecules, hydrogen atoms were fixed geometrically at calculated distances and allowed to ride on the parent non-hydrogen atoms with the isotropic displacement being fixed at 1.2 times of the atoms they attached to. Several solvent CHCl3 molecules were refined with the C–Cl bond distance being fixed at 1.57 Å and Cl⋯Cl separation of 2.50 Å, respectively. One of the uncoordinated nitrate anions was refined with the N–O bond distance being fixed at 1.25 Å and the separation between three oxygen atoms was fixed to be the same, respectively.
Results and discussion
Structural studies of Tb–PT1 and Sm–PT1
Ligand PT1 was synthesized according to the literature method.14 Layering a methanol solution of Sm(NO3)3·6H2O above the chloroform/methanol solution of PT1, like in case of the preparation of Tb–PT1 octahedron, led to the formation of molecular material Sm–PT1 with the yield of 62%. Single crystal structural analysis demonstrated the formation of the octahedron Sm–PT1, which was similar to the reported Eu–PT1 complex.14 Each octahedron comprised six metal ions positioned at the vertexes, and four ligands alternatively positioned on the four of the eight faces. The separation between two metal ions bridged by one ligand was about 10.5 Å, and the distance between two diagonal metal ions was about 14.9 Å, respectively, with the inner volume being estimated as 400 Å3. Each lanthanide ion was coordinated by two tridentate chelators from two different ligands and two nitrate anions, respectively. These nitrate anions were potentially removable, from which the lanthanide ions were possibly considered as coordinative unsaturated metal centers and potential acid-type catalytic active sites. These amide groups—located within the positively charged cages—provide static, geometric, coordinative, and functional properties to the cage-like capsules, which are important for the recognition of small biochemical molecules and for the base-type catalysis of special organic reactions.Importantly, the packing patterns of the two octahedral cages in their crystals were quite different (Scheme 1). Tb–PT1 crystallized in a space group Pc1. Each octahedron positioned at a crystallographic C3 axis and connected with three neighbors through intermolecular π⋯π stacking interactions, featuring a two-dimensional (6,3) sheet having a cavity of a radius of about 1.5 nm. Adjacent sheets were further packed into three-dimensional networks, exhibiting one-dimensional channels. Sm–PT1 crystallized in a space group R. The octahedron positioned at the crystallographic C3 axis also and connected with three neighbors through inter-molecular π⋯π interactions, featuring a 2D sheet similar to that of Tb–PT1. However, adjacent sheets in Sm–PT1 were packed in an ABAB fashion, blocking the 1D channels. In this case, the similar molecular structure but different packing patterns of Tb–PT1 and Sm–PT1 made the two compounds promising candidates to optimize the heterogeneous catalysis conditions of the given reactions. TGA analysis of crystalline samples Tb–PT1 and Sm–PT1 showed only 18% decrease in weight for both compounds at 210 °C, corresponding to the loss of the adsorbed solvents in the crystals. These results showed that both compounds were stable enough after losing the lattice solvent molecules and could be used as catalysts in a heterogeneous manner.
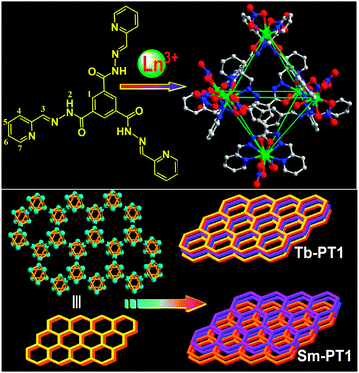 | ||
| Scheme 1 Structure of ligand PT1 and constructional fragments, showing the cationic lanthanide-based octahedral cages, and the packing diagram of the two dimensional layers consolidated by the octahedrons, showing the AAA fashion of the two-dimensional layers in the channel structure of Tb–PT1 and the ABAB fashion of the porous-like structure of Sm–PT1, respectively. | ||
Catalysis of cyanosilylation reaction of aldehydes
To understand the catalytic properties of these polyhedra and optimize the heterogeneous conditions of the given catalysts, cyanosilylation reaction provides a convenient route to cyanohydrins, which are key derivatives in the synthesis of fine chemicals and pharmaceuticals,18 and was investigated in a heterogeneous manner. As shown in Table 1, a loading of 1 mol% of Tb–PT1 (5 μmol) led to a 90% conversion of nitrobenzaldehydes after 1.5 h. The catalytic activity was comparable to the best prior results for MOFs.19 Solids of Tb–PT1 could be easily isolated from the reaction suspension by simple filtration alone and can be reused at least twice with a slight decrease in reactivity. Importantly, removal of Tb–PT1 by filtration after only 0.5 h completely shuts down the reaction, affording only 1.8% increase in conversion upon standing for 16 h. These results demonstrated that Tb–PT1 is a true heterogeneous catalyst. Furthermore, the conversion of 4-nitrobenzaldehyde (4-NBA) in a heterogeneous manner was just 11.8%, catalyzed by PT1 (4 mol% ratio) after 1.5 h under similar experimental conditions, and the reaction didn't progress without catalyst. It seemed that the lanthanide octahedra rather than the ligand acted as the dominant catalytic active sites.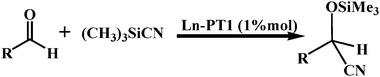
While the smooth reaction between 2-nitrobenzaldehyde (2-NBA) (molecular size, 5.8 × 5.3 Å2), 3-nitrobenzaldehyde (3-NBA) (molecular size, 6.9 × 5.0 Å2) or 4-NBA (molecular size, 7.0 × 4.3 Å2) and Me3SiCN suggested that the window size of the catalyst is larger enough to allow such kind of substrates to pass through, just 13% of the cyanosilylation product was observed when the reaction of bulky aldehyde with larger size, 3-formyl-1-phenylene-(3, 5-di-tert-butylbenzoate) (BA) (molecular size, 12.9 × 9.1 Å2),20 was conducted after the same period of time, in the presence of Tb–PT1 catalyst. Given the expectation of comparable reactivity, it seems that the diameter of the channel (11.5 Å eliminating the van der Waals radii of about 3.6 Å) is too small to readily accommodate the transition state geometry required for activating the corresponding substrates. The potential size-selectivity of the cyanosilylation reactions suggested that the catalytic reactions mostly occurred in the channels of the catalysts, not on the external surface.
The investigation on the adsorption of the substrates in the catalyst also confirmed the size-selectivity. The desolvated catalyst was immersed in the solid catalyst in a CH2Cl2 solution containing the substrates for 3 h, and then filtrated and washed by CH2Cl2 several times to remove the substrate remaining on the surface. 1H NMR (DMSO-d6) of the sample immersed in the CH2Cl2 solution containing 4-NBA exhibited peaks at chemical shifts of 10.18, 8.43 and 8.17 ppm, assigned to the proton signals of the substrate, all of them being shifted downfield (δ = 0.1 ppm) (Fig. 1a–c). Detailed 1H NMR spectral analyses demonstrated the 3:1 mole ratio of 4-NBA absorbed per polyhedron in the solid catalyst. However, 1H NMR (DMSO-d6) of Tb–PT1 immersed in a CH2Cl2 solution containing BA didn't exhibit any peaks of BA, which asserted that the BA was not incorporated by the catalyst (Fig. 1d).
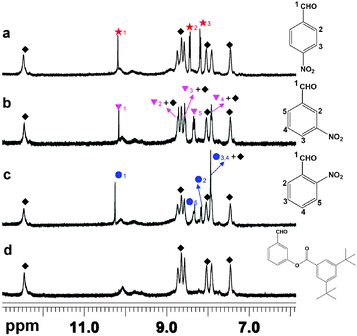 | ||
| Fig. 1 1H NMR (DMSO-d6) spectra of Tb–PT1 that adsorbed different aldehydes: (a) 4-NBA, (b) 3-NBA, (c) 2-NBA and (d) BA. The peaks marked with a black rhombus indicate the protons in Tb–PT1, the others refer to the substrates. | ||
To further probe whether activation of the carbonyl species occurs inside the channels or on the surface of the solid catalyst, fluorescent titrations of the suspension of Tb–PT1 in CH2Cl2 upon addition of the substrates were investigated. When excited at 365 nm, free Tb–PT1 exhibited two intense emission bands of 465 nm (PT1 emission) and 525 nm (the Tb3+ emission assignable to the transitions of 5D4 → 7F4).21 The addition of 4-NBA, 3-NBA and 2-NBA reduced the luminescence of the Tb3+ emission (525 nm) and PT1 emission (465 nm) of the suspension of Tb–PT1 in CH2Cl2, while both luminescence of the Tb3+ emission and PT1 emission of the suspension of Tb–PT1 were almost unaltered with the addition of BA. This result suggested that only the molecules having suitable sizes had the potential to enter the channels or be encapsulated by cavities, and interact with the luminescence sites from which the luminescent responses exhibited size-selective performance.
The quenching effect was rationalized by the Stern–Volmer equation:22
| I0/I = 1 + KSV [M] |
I 0 and I are the luminescence intensities of aldehyde-free Tb–PT1 and aldehyde-incorporated Tb–PT1, respectively, [M] is the molar concentration of the aldehydes added, and KSV is the quenching effect coefficient of the aldehyde. KSV was calculated to be ca. 5.4 M−1 from the experimental data for the examined aldehyde. The addition of 4-NBA to Tb–PT1 in an acetonitrile solution does not cause any obvious absorption spectroscopic changes, thus the fluorescence quenching by aldehydes should be ascribed to a PET (photo-induced energy transfer) mechanism between the guest molecules and Tb–PT1.23
Interestingly, KSV values referring to the intensities at 465 nm and at 525 nm are different (Fig. 2 inset). With the decrease of the intensities of the two emission bands upon incorporating 4-NBA, the I525/I465 ratio increased significantly. Since the two emission bands were isogenous, the characteristic Tb3+ emission was prompted by the excitation of the PT1 group. The quenching of the two emission bands was attributed to PET from the PT1 group to 4-NBA, whereas the increase of I525/I465 ratio should be attributed reasonably to the interactions between molecules of 4-NBA and the Tb3+ ions. Accordingly, the enhancement of the emitting efficiency at 525 nm (Tb3+ emission) was attributed to the replacement of the coordinated solvent molecules by the aldehydes (Fig. 3). These results demonstrated that the interactions corresponding to the Tb3+ sites were dominant in luminescent responses ratiometrically and in activating the aldehydes.
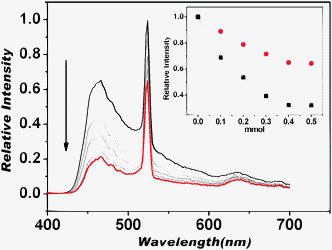 | ||
| Fig. 2 Family of luminescent spectra of Tb–PT1 emulsion in CH2Cl2 upon addition of various amounts of 4-NBA up to 0.5 mmol. The inset shows fluorescence responses of Tb–PT1 upon addition of 4-NBA in CH2Cl2 at 525 nm (red line) and at 465 nm (black line), excitation at 365 nm. | ||
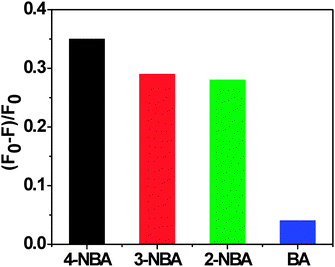 | ||
| Fig. 3 Luminescent responses of Tb–PT1 upon addition of aldehydes (0.5 mmol) of interest. Intensities were recorded at 525 nm, excitation at 365 nm. | ||
To further validate the catalytic reactions occurring within the channels and in the surface of the catalyst, another lanthanide-based porous material Sm–PT1 having the same molecular structure but a different packing pattern was investigated. Molecules of Sm–PT1 octahedron in the crystals were packed into two-dimensional layers, but the layers stacked in an ABAB fashion to block the channels into cavities. The sorption experiments including the 1H NMR (d6-DMSO) of the immersed sample suggested the same absorption of substrate 4-NBA within Sm–PT1, and revealed the presence of 3 mole equiv. of 4-NBA per polyhedron. The loading of only 1 mol% ratio of Sm–PT1 (5 μmol) led to excellent conversion of the nitrobenzaldehyde substrates, whereas the conversions of the corresponding reactions catalyzed by Sm–PT1 were lower compared to the reactions prompted by Tb–PT1. And the conversion of cyanosilylation with BA catalyzed by Sm–PT1 was the lowest one and almost the same as that by Tb–PT1, while the conversion of all the entries was over 97% using Tb(NO3)3 and Sm(NO3)3 as catalysts under the same reaction conditions, respectively. Because the two compounds have similar molecular structures and almost the same coordination geometry, the difference in the reaction conversions might be one of the indicators that the reactions occurred within the channels or pores of the catalysts. The higher conversion of the channel structure Tb–PT1 compared to that of the porous structure Sm–PT1 suggested that the presence of nanometre channels might benefit the nitrobenzaldehydes diffusing swiftly through the channels and accommodating the transition state geometry required for activating these substrates.
Catalyses of aldol reaction by Tb–PT1 and Sm–PT1
As mentioned in the literature, the amide groups seemed basic enough to abstract an α-H atom of a ketone to generate a nucleophilic enolate.24 Aldol reaction, one of the most powerful carbon–carbon bond-forming reactions,25 was chosen to optimize the heterogeneous catalytic conditions of these octahedra.26 As shown in Table 2, the loading of only 2 mol% ratio of Tb–PT1 (0.01 mmol) led to 80% conversion corresponding to 4-NBA and cyclohexanone with the diastereo-selectivity being 2:1 (syn:anti). And the catalysis efficiency of Tb–PT1 was comparable to the best prior result for metal–organic framework catalysts.27

| Entry | Substrate | Tb–PT1 | Sm–PT1 | PT1 | |||
|---|---|---|---|---|---|---|---|
| Con.% | Dr(s/a) | Con.% | Dr(s/a) | Con.% | Dr(s/a) | ||
| a The reaction was carried out at 37 °C for 5 d with cyclohexanone (5 mmol), aldehyde (0.5 mmol) and 0.01 mmol catalysts (2 mol%) in CH3OH/H2O, the conversion and diastereomer were determined by 1H NMR spectroscopy. | |||||||
| 5 | 4-NBA | 80.5 | 2.0:1 |
64.9 | 2.1:1 |
4.8 | syn |
| 6 | 3-NBA | 75.2 | 1.7:1 |
56.0 | 2.3:1 |
8.9 | syn |
| 7 | 2-NBA | 48.2 | 2.1:1 |
31.5 | 1.4:1 |
8.3 | syn |
| 8 | BA | 27.5 | 2.4:1 |
21.3 | 2.4:1 |
9.1 | syn |
Furthermore, in the heterogeneous manner the conversion of 4-NBA and cyclohexanone was just 4.8% with only cis isomer catalyzed by PT1 (8 mol% ratio) under the same conditions. Tb–PT1 solids could also be easily isolated from the reaction suspension by simple filtration alone and can be reused at least twice with a slight decrease in reactivity. Whereas the conversions of other two nitrobenzaldehydes with different dimensions (75.2% and 48.2%) were much lower than 4-NBA prompted by Tb–PT1, only 27.5% of the aldol product was observed in the case of the bulky aldehyde BA as substrate after the same period of time in the presence of Tb–PT1. While the sorption investigation and the catalytic cyanosilylation reactions of various aldehydes with the crystalline solids of Tb–PT1 suggested that molecules of all the three nitrobenzaldehydes could smoothly pass through the channels of the catalysts. The lower catalytic effect in the case of 2-, or 3- nitrobenzaldehydes as substrates should be likely attributed to that the size of the opening regular trigon within the octahedral molecule is too small for them to enter the inner cavities of the octahedra themselves, even considering the possible difference in their reactive activity. Accordingly, we think that the aldol reactions should take place within the octahedral cavities, and distinguish conversions rooted in the size of the opening regular trigon having an edge of about 10 Å2 within the octahedral molecule.
To further validate the potential catalytic mechanism and probe whether activation of the carbonyl species occurs inside the inner cavities of the polyhedron or in the channels/on the surface of the solid catalyst, fluorescent titration of the suspension of Tb–PT1 in CH2Cl2 upon addition of the cyclohexanone was investigated. As mentioned above, the Uv-vis spectra of Tb–PT1 do not have any significant absorption variation upon addition of 4-NBA even at high concentration. However, the addition of cyclohexanone led to the significant absorbance decrease at the peaks 330 nm and 365 nm, and caused the appearance of a new peak at about 415 nm (Fig. 4). The presence of sharp isosbestic points at about 400 nm and 350 nm indicates that only two species coexist in the equilibrium. Since the bands at 330 nm and 415 nm were mainly attributed to the ligand-based charge transfer bands,14 the absorption variations of the catalyst were mainly attributed to the potential interactions corresponding to the amide group. Importantly, the addition of cyclohexanone to the suspension of Tb–PT1 in CH2Cl2 caused luminescence enhancement of the Tb3+ emission (525 nm) and ligand-based emission (465 nm) significantly (Fig. 5). As the two bands were isogenous, and the characteristic Tb3+ emission was prompted by the excitation of the PT1 group, the luminescent enhancements of the two emission bands were reasonably attributed to interactions between the cyclohexanone and the amide groups in the ligand backbone. Clearly, these results revealed that the interactions between cyclohexanone and the solid of the catalyst were mainly taking place at the active sites on the ligand backbone, mainly attributed to the potential hydrogen bonding between the cyclohexanone molecules and amide groups.
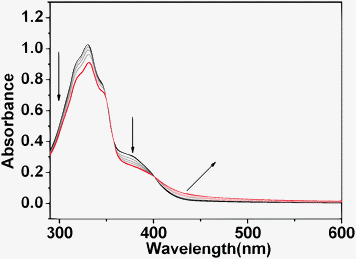 | ||
| Fig. 4 Uv-vis spectra of the Tb–PT1 (5 × 10−6 M) upon addition of cyclohexanone (0.25 mM) to an acetontrile solution (a); Luminescent responses of Tb–PT1 upon addition of cyclohexanone (0.1 mL to 0.5 mL). | ||
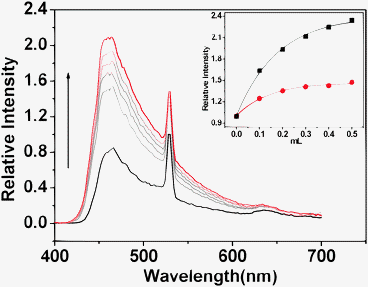 | ||
| Fig. 5 Fluorescence responses of Tb–PT1 upon addition of cyclohexanone in CH2Cl2 at 525 nm (red line) and at 465 nm (black line). Excitation at 365 nm (inset). | ||
Interestingly, under the same experimental conditions, the loading of only 2 mol% ratio of Sm–PT1 (0.01 mmol) catalyst led to 65% conversion corresponding to 4-NBA with the diastereoselectivity of 2:1 (syn:anti). The 21.3%/27.5% conversion (entry 8) in the presence of Sm–PT1/Tb–PT1 suggested that the narrow triangle opening of these molecular polyhedra was too small for BA to pass through. The almost same conversion and the same size-selective sequence of the two catalysts confirmed that the aldol reactions should take place within the octahedral cavities, given that the two compounds have similar molecular structures and almost the same coordination geometry, but different packing patterns. While the immobilization of unsaturated Lewis acid Ln ions within the relatively open frameworks having well-defined pores represents extensively investigated methodologies and enables researchers to target some unique porous LnMOFs with multifunctional properties and catalytic applications.28 The heterogeneous catalytic behavior of our materials is quite significant, it not only suggests that the porous molecular materials comprised of semi-infinite macrocycles have the potential to combine the excellent size discrimination properties of the macrocycles in solution and the advantage of the heterogeneous catalysts, but also demonstrates that the catalysts containing unsaturated lanthanide ions positioned at the intermolecular cavities of the crystals and amide groups within the inner cavities of the polyhedron have the potential to exhibit both base-type and acid-type catalytic driving forces directly. And owing to the different catalytic mechanisms, our systems also provide the possibility to tune the size-selectivity through controlling the catalytic reactions within the inner cavities of the discrete molecular polyhedron or within the intermolecular channels or pores of the materials consolidated.
The as-synthesized crystals of polyhedra lost part of their lattice molecules under ambient conditions. However, when the desolvated amorphous powders were reacted, they were restored to the original crystalline phase, which was detected by the XRPD patterns. Crystalline solids of the two compounds were also easily isolated from the reaction suspension by filtration alone and can be reused at least twice without loss of activity. The maintenance of the crystalline information of the solid compounds during the reaction processes suggested the possibility of the powder to be recycled with a comparable catalytic activity.
Conclusions
In summary, we reported the catalytic properties of multifunctional lanthanide–organic octahedra Tb–PT1 and Sm–PT1 featuring both Lewis acidic Ln3+ sites and basic amide group sites. These molecular materials catalyze the cyanosilylation and aldol reactions in a size-selective fashion through base-type and acid-type catalysis sites, respectively. Crystalline solids of the catalysts were easily isolated from the reaction suspension by filtration alone and can be reused without loss of activity, suggesting that the catalysts were recycled. The size-selective behaviors coupled with the guest–host spectroscopic investigation between the reaction substrates and the catalyst suggested that the cyanosilylation reactions mostly took part in the channel of the catalysts and the aldehydes substrates were activated by the Ln3+ ions, while the aldol reactions mainly occurred in the octahedral cavities and the cyclohexanone substrates possibly interacted with the amide groups through a hydrogen bond, respectively. These crystalline molecular materials hierarchically assembled from semi-infinite metal–organic polyhedra are thus expected to be useful catalysts which could combine the advantage of a heterogeneous catalyst and the excellent size discrimination properties of the polyhedra in solution. The efficient approach will be extended to other lanthanide-based polyhedra systems having both coordination unsaturated metal centers and a series of organic active sites to hierarchically assemble more efficient acid- and/or base-type porous heterogeneous catalysts of organic synthetic interest.Acknowledgements
This work is supported by the National Natural Science Foundation of China (20801008).Notes and references
- (a) B. Olenyuk, J. A. Whiteford and P. J. Stang, Nature, 1999, 398, 796–799 CrossRef CAS; (b) N. Takeda, K. Umemoto, K. Yamaguchi and M. Fujita, Nature, 1999, 398, 794–796 CrossRef CAS; (c) D. J. Tranchemontagne, Z. Ni, M. O'Keeffe and O. M. Yaghi, Angew. Chem., Int. Ed., 2008, 47, 5136–5147 CrossRef CAS; (d) J.-R. Li and H.-C. Zhou, Nat. Chem., 2010, 2, 893 CrossRef CAS.
- (a) F. Hof and J. Rebek, Jr., Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4775–4777 CrossRef CAS; (b) M. D. Pluth, R. G. Bergman and K. N. Raymond, Science, 2007, 316, 85–88 CrossRef CAS; (c) M. Jung, H. Kim, K. Baek and K. Kim, Angew. Chem., Int. Ed., 2008, 47, 5755–5757 CrossRef CAS; (d) T. K. Ronson, J. Fisher, L. P. Harding, P. J. Rizkallah, J. E. Warren and M. J. Hardie, Nat. Chem., 2009, 1, 212–216 CrossRef CAS.
- (a) D. L. Caulder and K. N. Raymond, Acc. Chem. Res., 1999, 32, 975–982 CrossRef CAS; (b) S. Leininger, B. Olenyuk and P. J. Stang, Chem. Rev., 2000, 100, 853–908 CrossRef CAS; (c) M. Fujita, K. Umemoto, M. Yoshizawa, N. Fujita, T. Kusukawa and K. Biradha, Chem. Commun., 2001, 509–518 RSC; (d) R. M. McKinlay, G. W. V. Cave and J. L. Atwood, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 5944–5948 CrossRef CAS.
- (a) N. C. Gianneschi, M. S. Maser and C. A. Mirkin, Acc. Chem. Res., 2005, 38, 825–837 CrossRef CAS; (b) T. D. Hamilton, G. S. Papaefstathiou, T. Friscic, D. K. Bucar and L. R. MacGillivray, J. Am. Chem. Soc., 2008, 130, 14366–14367 CrossRef CAS.
- (a) P. J. Stang and B. Olenyuk, Acc. Chem. Res., 1997, 30, 502–518 CrossRef CAS; (b) S. R. Seidel and P. J. Stang, Acc. Chem. Res., 2002, 35, 972–983 CrossRef CAS; (c) S. J. Dalgarno, N. P. Power and J. L. Atwood, Coord. Chem. Rev., 2008, 252, 825–841 CrossRef CAS; (d) M. Yoshizawa, J. K. Klosterman and M. Fujita, Angew. Chem., Int. Ed., 2009, 48, 3418–3438 CrossRef CAS.
- (a) D. L. Caulder and R. N. Raymond, Acc. Chem. Res., 1999, 32, 975–982 CrossRef CAS; (b) D. Fiedler, D. H. Leung, R. G. Bergman and K. N. Raymond, Acc. Chem. Res., 2005, 38, 349–358 CrossRef CAS; (c) R. W. Saalfrank, H. Maid and A. Scheurer, Angew. Chem., Int. Ed., 2008, 47, 8794–8824 CrossRef CAS; (d) P. Mal, B. Breiner, K. Rissanen and J. R. Nitschke, Science, 2009, 324, 1697–1699 CrossRef CAS.
- M. D. Pluth, R. G. Bergman and K. N. Raymond, Science, 2007, 316, 85–88 CrossRef CAS.
- M. Yoshizawa, M. Tamura and M. Fujita, Science, 2006, 312, 251–254 CrossRef CAS.
- (a) J. Meeuwissen and J. N. H. Reek, Nat. Chem., 2010, 2, 615–621 CrossRef CAS; (b) T. S. Koblenz, J. Wassenaar and J. N. H. Reek, Chem. Soc. Rev., 2008, 37, 247–262 RSC; (c) P. W. N. M. Van Leeuwen, Supramolecular Catalysis, Wiley-VCH, 2008 Search PubMed.
- (a) M. Heitbaum, F. Glorius and I. Escher, Angew. Chem., Int. Ed., 2006, 45, 4732 CrossRef CAS; (b) Principles and Practice of Heterogeneous Catalysis, ed. J. M. Thomas and W. J. Thomas, Wiley-VCH, Weinheim, 1997 Search PubMed; (c) H.-U. Blaser, B. Pugin and M. Studer, in Chiral Catalyst Immobilization and Recycling, ed. D. E. DeVos, I. F. J. Vankelecom and P. A. Jacobs, Wiley-VCH, Weinheim, 2000, p. 1 Search PubMed.
- (a) J. Y. Lee, O. K. Farha, J. K. Roberts, A. Scheidt, S. B. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450–1459 RSC; (b) L. Q. Ma, C. Abney and W. B. Lin, Chem. Soc. Rev., 2009, 38, 1248–1256 RSC.
- (a) J. I. García, B. López-Sánchez and J. A. Mayoral, Org. Lett., 2008, 10, 4995–4998 CrossRef; (b) S. H. Cho, B. Q. Ma, S. T. Nguyen, J. T. Hupp and T. E. Albrecht-Schmitt, Chem. Commun., 2006, 2563–2564 RSC.
- (a) V. Chandrasekhar, P. Thilagar, J. F. Bickley and A. Steiner, J. Am. Chem. Soc., 2005, 127, 11556–11557 CrossRef CAS; (b) G. Li, W.-B. Yu and Y. Cui, J. Am. Chem. Soc., 2008, 130, 4582–4583 CrossRef CAS; (c) Y. Liu, R. Zhang, C. He, D. B. Dang and C. Y. Duan, Chem. Commun., 2010, 46, 746–748 RSC.
- C. He, Z. H. Lin, Z. He, C. Y. Duan, C. H. Xu, Z. M. Wang and C. H. Yan, Angew. Chem., Int. Ed., 2008, 47, 877–881 CrossRef CAS.
- D. Moon, S. Kang, J. Park, K. Lee, R. P. John, H. Won, G. H. Seong, Y. S. Kim, G. H. Kim, H. Rhee and M. S. Lah, J. Am. Chem. Soc., 2006, 128, 3530–3531 CrossRef CAS.
- (a) S. Hasegawa, S. Horike, R. Matsuda, S. Furukawa, K. Mochizuki, Y. Kinoshita and S. Kitagawa, J. Am. Chem. Soc., 2007, 129, 2607–2614 CrossRef CAS; (b) Y. Zou, M. Park, S. Hong and M. S. Lah, Chem. Commun., 2008, 2340–2342 RSC.
- (a) X. Xu, M. Ma, Y. Yao, Y. Zhang and Q. Shen, Eur. J. Inorg. Chem., 2005, 676–684 CrossRef; (b) T. N. Parac-Vogt, S. Pachini, P. Nockemann, K. V. Hecke, L. V. Meervelt and K. Binnemans, Eur. J. Org. Chem., 2004, 4560–4566 CrossRef CAS.
- (a) K. Higuchi, M. Onaka and Y. Izumi, Bull. Chem. Soc. Jpn., 1993, 66, 2016–2032 CrossRef CAS; (b) R. J. H. Gregory, Chem. Rev., 1999, 99, 3649–3682 CrossRef CAS; (c) M. North, Tetrahedron: Asymmetry, 2003, 14, 147–176 CrossRef CAS.
- (a) M. Fujita, Y. J. Kwon, S. Washizu and K. Ogura, J. Am. Chem. Soc., 1994, 116, 1151–1152 CrossRef CAS; (b) O. R. Evans, H. L. Ngo and W. B. Lin, J. Am. Chem. Soc., 2001, 123, 10395–10396 CrossRef CAS; (c) K. Schlichte, T. Kratzke and S. Kaskel, Microporous Mesoporous Mater., 2004, 73, 81–88 CrossRef CAS; (d) S. Horike, M. Dinca, K. Tamaki and J. R. Long, J. Am. Chem. Soc., 2008, 130, 5854–5855 CrossRef CAS; (e) S. Neogi, M. K. Sharma and P. K. Bharadwaj, J. Mol. Catal. A: Chem., 2009, 299, 1–4 CrossRef CAS.
- These molecular sizes are calculated based on the Chem Office soft.
- (a) Z. Li, G. Zhu, X. Guo, X. Zhao, Z. Jin and S. Qiu, Inorg. Chem., 2007, 46, 5174–5178 CrossRef CAS; (b) B. Zhao, X. Y. Chen, P. Cheng, D. Z. Liao, S. P. Yan and Z. H. Jiang, J. Am. Chem. Soc., 2004, 126, 15394–15395 CrossRef CAS; (c) B. Chen, L. Wang, F. Zapata, G. Qian and E. B. Lobkovsky, J. Am. Chem. Soc., 2008, 130, 6718–6719 CrossRef CAS; (d) K.-L. Wong, G.-L. Law, Y.-Y. Yang and W.-T. Wong, Adv. Mater., 2006, 18, 1051–1054 CrossRef CAS.
- (a) Y. Xiao, Y. Cui, Q. Zheng, S. Xiang, G. Qian and B. Chen, Chem. Commun., 2010, 46, 5503–5505 RSC; (b) B. Chen, L. Wang, Y. Xiao, F. R. Fronczek, M. Xue, Y. Cui and G. Qian, Angew. Chem., Int. Ed., 2009, 48, 500–503 CrossRef CAS.
- (a) P. Jiang and Z. Guo, Coord. Chem. Rev., 2004, 248, 205–229 CrossRef CAS; (b) R. Kramer, Angew. Chem., Int. Ed., 1998, 37, 772–773 CrossRef CAS; (c) L. Fabbrizzi, M. Licchelli, P. Pallavicini, A. Perotti, A. Taglietti and D. Sacchi, Chem.–Eur. J., 1996, 2, 75–82 CrossRef CAS; (d) M. Boiocchi, L. Fabbrizzi, M. Licchelli, D. Sacchi, M. Vazquez and C. Zampa, Chem. Commun., 2003, 1812–1813 RSC.
- (a) C. M. Lee and W. D. Kumler, J. Am. Chem. Soc., 1962, 84, 571–578 CrossRef CAS; (b) H. A. Bent, Chem. Rev., 1968, 68, 587–648 CrossRef CAS; (c) P. L. Huyskens, J. Am. Chem. Soc., 1977, 99, 2578–2582 CrossRef CAS.
- (a) L. G. Wade, Organic Chemistry, Prentice Hall, Upper Saddle River, New Jersey, 6th edn, 2005, pp. 1056–1066 Search PubMed; (b) M. B. Smith and J. March, Advanced Organic Chemistry, Wiley Interscience, New York, 5th edn, 2001, pp. 1218–1223 Search PubMed; (c) R. Mahrwald, Modern Aldol Reactions, Wiley-VCH Verlag GmbH & Co. KGaA., Weinheim, Germany, 2004, vol. 1 and 2, pp. 1218–1223 Search PubMed.
- (a) A. Olmos, A. Alix, J. Sommer and P. Pale, Chem.–Eur. J., 2009, 15, 11229–11234 CrossRef CAS; (b) S. Luo, J. Li, L. Zhang, H. Xu and J. P. Cheng, Chem.–Eur. J., 2008, 14, 1273–1281 CrossRef CAS.
- (a) M. Banerjee, S. Das, M. Yoon, H. J. Choi, M. H. Hyun, S. M. Park, G. Seo and K. Kim, J. Am. Chem. Soc., 2009, 131, 7524–7525 CrossRef CAS; (b) S. K. Ghosh, S. Bureekaew and S. Kitagawa, Angew. Chem., Int. Ed., 2008, 47, 3403–3406 CrossRef CAS.
- (a) M. Gustafsson, A. Bartoszewicz, B. Martin-Matute, J. L. Sun, J. Grins, T. Zhao, Z. Y. Li, G. S. Zhu and X. D. Zou, Chem. Mater., 2010, 22, 3316–3322 CrossRef CAS; (b) S. Kitagawa, S. I. Noro and T. Nakamura, Chem. Commun., 2006, 701–707 RSC; (c) O. R. Evans, H. L. Ngo and W. Lin, J. Am. Chem. Soc., 2001, 123, 10395–10396 CrossRef CAS; (d) N. Snejko, C. Cascales, B. Gomez-Lor, E. Gutiérrez-Puebla, M. Iglesias, C. Ruiz-Valero and M. A. Monge, Chem. Commun., 2002, 1366–1367 RSC; (e) J. W. Han and C. L. Hill, J. Am. Chem. Soc., 2007, 129, 15094–15095 CrossRef CAS; (f) T. Dewa, T. Saiki and Y. Aoyama, J. Am. Chem. Soc., 2001, 123, 502–503 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 675067. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1nj20736a |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2012 |