Carbeniophosphanes and their carbon → phosphorus → metal ternary complexes
Yves
Canac
*ab,
Carine
Maaliki
ab,
Ibrahim
Abdellah
ab and
Remi
Chauvin
*ab
aCNRS, LCC (Laboratoire de Chimie de Coordination), 205, route de Narbonne, F-31077 Toulouse, France. E-mail: yves.canac@lcc-toulouse.fr; chauvin@lcc-toulouse.fr; Fax: (+33)5 61 55 30 03
bUniversité de Toulouse, UPS, INP, LCC, F-31077 Toulouse, France
First published on 25th November 2011
Abstract
The discovery, recent developments and prospects of the carbeniophosphines, and in particular of imidazoliophosphines, are related. The P-conjugated positive charge brought by the stabilized carbenium center gives stable (Im+)R2P → metal dative bonds, while the ligands themselves contain a relatively stable C → P+R2 dative bond, making imidazoliophosphines, more accurately described as NHC–phosphenium adducts. The preparation methods, structural features, electronic and coordination properties, reactivity, and catalytic properties of carbeniophosphines are reviewed.
 Yves Canac | Yves Canac obtained his PhD degree in 1996 under the supervision of Prof. Guy Bertrand in Toulouse (France). After a postdoctoral fellowship with Prof. Andrea Vasella at the ETH-Zürich (Switzerland), he entered the CNRS in 1998 at the University of Paris XI-Orsay (France). In 2001, he moved to the University of California-Riverside (UCR), taking part in the foundation of the joint Research Chemistry Laboratory UCR/CNRS. In 2006, he came back to Toulouse at the CNRS Laboratory of Coordination Chemistry. His research activities are mainly focused on the design, the coordination chemistry and the catalytic properties of novel chiral electron-rich carbon ligands (NHCs, ylides) and electron-poor cationic phosphorus ligands (amidiniophosphanes). |
 Carine Maaliki | Carine Maaliki, born in 1984, obtained her Master's degree in Chemistry in 2009 from the University Paul-Sabatier in Toulouse (France), with specialization in therapeutic chemistry. She is currently working as a PhD student at the “Laboratoire de Chimie de Coordination du CNRS” in Toulouse, under the co-supervision of Prof R. Chauvin and Dr Y. Canac. Her scientific activity relates to the design, the preparation and the coordination chemistry of cationic phosphorus ligands (amidiniophosphines) for catalytic applications. |
 Ibrahim Abdellah | Ibrahim ABDELLAH received his Master degree from the University of Paris-Est Créteil (France) in 2007. Then, he obtained his PhD degree under the supervision of Prof. Remi Chauvin and Dr Yves Canac at the “Laboratoire de Chimie de Coordination du CNRS” in Toulouse (France) in 2010, on the synthesis and coordination chemistry of carbon (NHCs, ylides) and phosphorus (amidiniophosphines) chiral ligands with extreme donating character. He is currently a postdoctoral fellow in the group of Prof. Annie-Claude Gaumont at the “Laboratoire de Chimie Moléculaire et Thio-organique—ENSICAEN” in Caen (France), working in the area of copper catalyzed carbon–phosphorus cross-coupling reactions. |
 Remi Chauvin | Remi Chauvin received his PhD in 1988 under the supervision of Prof. H. Kagan in Orsay (France). He continued as a post-doctoral fellow with K. B. Sharpless at MIT (USA), and then with A. Vasella in Zürich (Switzerland). On his return to France, he first worked at the Roussel Uclaf company in Romainville. In 1993 he came back to academic research at the CNRS in Toulouse, and in 1998 was appointed as a Professor at the Paul Sabatier University. Today he is interested in four aspects of molecular chemistry in two different laboratories: the Laboratory of Coordination Chemistry, where his main research group is hosted, and the Laboratory of Molecules of Biological Interest. The corresponding domains are theoretical analysis (molecular modeling, aromaticity,…), acetylenic synthesis (carbo-mer chemistry), coordination chemistry and catalysis (chiral carbon–phosphorus ligands), and synthesis of biologically active products (natural polyyne-polyols and analogues). |
1. Introduction
Wilkinson's discovery of the catalytic properties of rhodium and ruthenium complexes of triphenylphosphine1 has initiated a forty-year period of extremely active research on the design of phosphorus ligands of transition metals. Hundreds of possibly chiral N-, O-, or S-functional phosphines and di-or triphosphines with various backbones and coordinating abilities have thus been devised for specific applications in catalysis.2 In spite of the performance of analogous group 15 ligands (amines and arsines),3 it became soon evident that a “revolution” in this field had to rely on a more drastic change of the basic nature of the coordinating center. And indeed, the revival came through the change of the P(III) by C(II) coordinating atoms, with the advent of a class of stable singlet carbene ligands, giving strong C → metal dative bonds, namely the N-heterocyclic carbenes (NHCs) and, more particularly, the cyclic diamino-carbenes.4 These ligands are actually specifically competing with and often overcoming the alkylphosphines for their σ-donating properties.5 Other types of even more σ-donating carbon-based ligands were later devised or identified, such as non diamino-carbenes,6ylide-type carbanions (phosphonium ylides and carbodiphosphoranes),7 or still bent allenes.6a Nevertheless, behind the efforts devoted to the design of such electron-rich ligands, the search for electron-poor counterparts8 remains desirable for catalytic processes where the Lewis acidity of the metallic center is crucial.9 From a more general viewpoint, the design of novel ligands that push both upward and downward on the electronic parameter scale is also of fundamental interest. Until recently, electron-poor ligands have been mainly represented by phosph(in)ites, where the electron-withdrawing effects result from π-retro-donation and σ-(−I)-induction through oxygen linkages.10 The main limitation of these ligands is, however, the lability of the P–O bonds (in particular their hydrolytic sensitivity), and the design of robust ligands that would mimic phosphites is therefore challenging. Along this line, fluoroarylphosphines containing relatively inert P–C bonds constitute an alternative,11 but present limited structural diversity with remote C–F functionalities. More electronegative than fluorine atoms are, however, cationic centers. In most cationic phosphane ligands, the ammonium,12imidazolium13 or phosphonium14 centers have been introduced in a remote position from the coordinating P(III) atoms with the aim of designing ‘true’ zwitterionic organometallates,15 and/or performing catalysis in aqueous or polar media without altering the electronic properties of the active metal center.16 In the opposite prospect of making the metal center less electron-rich on purpose, the key is to introduce the cationic center in close proximity to the coordinating atom. This is achieved in amidiniophosphanes, where a stabilized carbenium center, namely a diaminocarbenium N2C+, is directly bonded to a P(III) atom. In the cyclic version, where a NHC unit was earlier assumed to occur as a coordinative motif, it was only recently demonstrated that imidazoliophosphanes are more accurately described as donor–acceptor NHC–phosphenium adducts.17 As highlighted below, the corresponding P(III) lone pair is not too far delocalized over the amidinium moiety and remains available for coordination to various metal fragments (Scheme 1).18 The corresponding carbeniophosphine–metal complexes (in the classical donor–acceptor binary description) should therefore be regarded as sequential ternary carbene–phosphenium–metal complexes involving two successive dative bonds (Scheme 1). Given a chemical bond A–B between two fragments of a representative Lewis structure (with a minimum number of formal atomic charges on A and B), three types of dissociation energies can be a priori calculated in the gas phase (e.g. by DFT methods): that of the homolytic process A–B → A˙ + ˙B (D0), and those of the heterolytic processes A–B → A+ + :B−, (D+) and A–B → A:− + B+ (D−). The bond is dative if the heterolytic mode is favored (i.e. D+ or D− < D0) allowing one to consider the Lewis structure of the complex as a directed chemical graph according to the sign of D+ − D−. The “dative degree” of the complex is the number of atoms involved in cumulated dative bonds. “Binary dative” complexes are thus those where the dative bonds are isolated, and two types of ternary dative complexes can be distinguished: the classical central complexes A → C ← B, and the sequential complexes A → B → C (Scheme 1).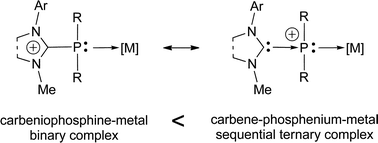 | ||
| Scheme 1 A mesomeric description of amidiniophosphine metal complexes: the sequential ternary form is the dominating one. | ||
This particular bonding situation combines the traditional P(III) and C(II) coordinative veins of phosphanes and NHCs, respectively (see above), and thus deserves particular attention. Moreover, the cationic nature of the carbeniophosphane ligands, and more specifically the dicationic nature of their bidentate versions, addresses the more general question of the role of electrostatic interactions in organometallic complexes, in particular regarding their catalytic properties.19
These original features are illustrated below by selected recent advances in the chemistry of carbeniophosphanes. Their preparation methods, electronic and coordinating properties, intrinsic reactivity, and the catalytic properties of their transition metal complexes are addressed. In spite of the energetic-structural information summarized in Scheme 1, the topological C+–P Lewis notation correponding to the “carbeniophosphane” designation will be adopted throughout the schemes (the dative C: → P+ notation is not strictly a Lewis symbol).
2. Preparation of carbeniophosphanes
2.1. Imidazoliophosphine systems
Most carbeniophosphanes known to date are imidazoliophosphines, which were generated by phosphinylation of the corresponding free NHC or its mesoionic imidazoliocarboxylate equivalent (Scheme 2). After a brief mention of an imidazoliophosphine by Zoller in 1988,20 the first fully characterized example was reported by Kuhn et al. ten years later. A representative 5 was thus obtained in good yield by reaction of the corresponding imidazol-2-ylidene 1 with chlorodiphenylphosphine (Scheme 2, route A).21 In the same spirit, Andrieu et al. described a large library of imidazoliophosphines 5 generated by reaction of the corresponding 1,3-dimethylimidazolium-carboxylate NHC equivalents 2 with various chlorophosphines (Scheme 2, route B).22 The process could be regarded as a displacement of the Cl− ligand of phosphenium chlorides (Cl–PR2 ↔ Cl−, [PR2]+) by NHCs.23 Very recently, a NHC–dichlorophosphenium adduct 5 was prepared by reaction of PCl3 with the 2-silylimidazolium salt 3 as an “onio-substituent transfer” reagent itself prepared by the reaction of the corresponding free NHC with Me3SiOTf (Scheme 2, route C).24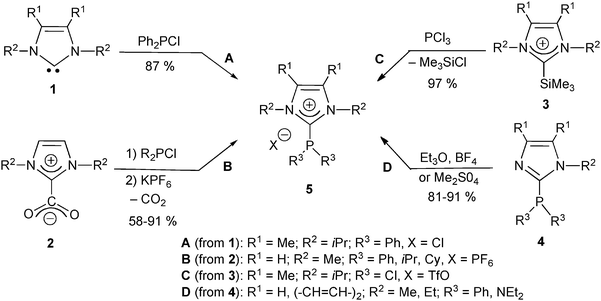 | ||
| Scheme 2 Alternative routes to generic NHC–phospheniums 5: (A) from imidazol-2-ylidenes;21 (B) from 2-carboxyimidazolium salts;22 (C) from 2-silylimidazolium salts;24 (D) from 2-phosphinoimidazoles.25 | ||
In a different approach circumventing the generation of highly reactive carbene nucleophiles, similar carbeniophosphine targets 5 can also be obtained by selective N-alkylation of neutral imidazolophosphine precursors 4 (route D).25 This strategy was recently applied for the synthesis of the phosphino-imidazoliophosphines 6b (BIMIONAP) and 7b and the bis(imidazoliophosphine) 7c from the corresponding BIMINAP26 and 1,2-bis(N-diphenylphosphinoimidazolyl)benzene27 precursors 6a and 7a, respectively (Scheme 3).
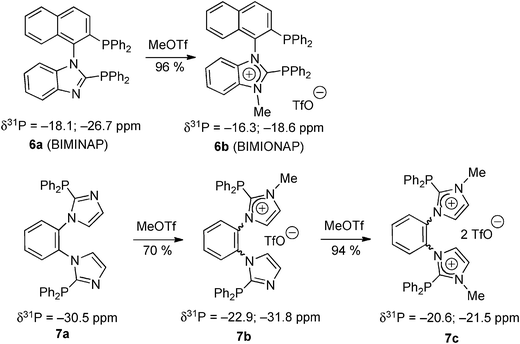 | ||
| Scheme 3 The synthesis of phosphino-imidazoliophosphines and a bis(imidazoliophosphine) by N-methylation of the imidazolophosphine precursors.26,27 | ||
The N-alkylation route can also be used even if the phosphane is coordinated beforehand to a sufficiently electron-rich metal fragment, like in the tungsten(0) complex 8a (Scheme 4),28 where the W(CO)5 unit plays the auxiliary role of a protecting group of the diphenylphosphino motif.
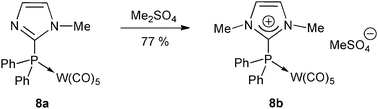 | ||
| Scheme 4 N-Methylation of the neutral tungsten(0) complex 8a to the carbeniophosphine complex 8b.28 | ||
Following the original method (Scheme 2, route A), a phosphiranylium cation stabilized by NHC-coordination 10 was recently described. It was obtained by displacement of the intramolecular tertiary amine ligand of the phosphiranylium complex 9 by the independently generated NHC, acting as a stronger donor than the amine (Scheme 5).29 It is noteworthy that both 9 and 10 can also be described by their amoniophosphine and carbeniophosphine Lewis structures, respectively. DFT studies of 10 confirmed the strong σ-donation of the NHC lone pair to the empty p orbital of the phosphenium center, but revealed a small π-back-donation of the phosphenium lone pair competing with the nitrogen lone pairs in their interaction with the empty pz* orbital of the sp2-carbon atom.
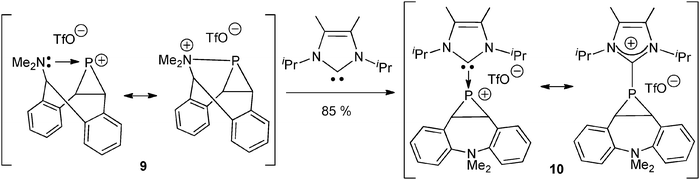 | ||
| Scheme 5 The preparation of the NHC–phosphiranylium adduct 10 from the intramolecular amine–phosphiranylium complex 9.29 | ||
2.2. Non-imidazolio carbeniophosphine systems
An alternative preparative method was disclosed by Alcarazo et al. for the synthesis of non-NHC carbene–phosphenium complexes.30 The cyclopropenylylidene–phosphenium adducts 12a–e were thus obtained in high yields by reaction of the chlorocyclopropenium salt 11 with secondary phosphines (Scheme 6). This result could be related to previous reports by Buford et al. on generic donor–phosphenium adducts exemplified by phosphine donors.31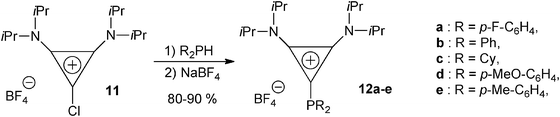 | ||
| Scheme 6 The synthesis of diaminocyclopropenylylidene–phosphenium complexes 12a–e from chlorodiaminocyclopropenium 11 and secondary diaryl- and dialkyl-phosphines.30 | ||
3. Electronic vs. coordinative properties of carbeniophosphanes
3.1. Nickel–carbonyl complexes
As argued in the introduction, the presence of a positive charge conjugated to the P(III) center is expected to make amidiniophosphanes weakly σ-donating ligands. It was, however, shown that they retain sufficient donating character for coordinating a metallic center. To quantify the overall σ-donating vs. π-accepting properties, the values of the IR stretching frequencies of carbonyl co-ligands at the metal atom were thus determined. For comparison with the large database initiated by Tolman with the Ni(CO)3 reference center, Andrieu et al. prepared nickel–carbonyl complexes 14 of various P-substituted amidiniophosphines 13 (Scheme 7).22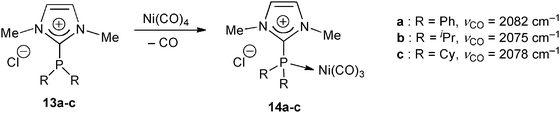 | ||
| Scheme 7 Nickel–carbonyl complexes 14 as probes of the σ-donating vs. π-accepting character of the imidazoliophosphane ligands 13.22 | ||
The characteristic νCO(A1) frequency of 14a at 2082 cm−1 thus allowed the amidiniophosphane 13a to be classified as less electron-donating than triphenylphosphine (νCO(A1) = 2069 cm−1). As expected from the stronger +I inductive effect of alkyl groups, replacement of the phenyl substituents by isopropyl and cyclohexyl counterparts induced a shift to lower wave numbers at 2075 and 2078 cm−1, respectively. The overall donating character of the imidazoliophosphines 13a–c was actually found in the range of the π-accepting phosphite references [P(OiPr)3: 2076 cm−1; P(OPh)3: 2085 cm−1],32 supporting that they could behave as real alternatives to phosphites for catalytic processes requiring electron-poor ligands.
3.2. Rhodium–carbonyl complexes
The single νCO stretching frequency of RhCl(CO) complexes is also currently used for estimating the donor character of co-ligands.33 The rhodium carbonyl complexes 15 and 16 were thus prepared from the carbeniophosphanes 5 (Scheme 2, R1 = H, R2 = Me, R3 = Ph)34 and 7c (Scheme 3),27 respectively (Scheme 8). The corresponding νCO values at 2003 and 2007 cm−1 are also close to that reported for the analogous phosphite complex trans-RhCl(CO)[P(OMe)3]2 (2011 cm−1, Scheme 8).35 In the chelate complex 16 (=(trans-η2-7c)RhCl(CO)2), the dicationic nature of the ligand 7c has thus no influence on the donor ability of the imidazoliophosphine moieties, showing that the o-phenylene bridge is electronically insulating. On the other hand, this semi-rigid link is also sufficiently large for permitting a trans-coordinating mode in the nine-membered rhodacycle of 16, thus allowing for a direct comparison with complex 15. Whereas the trans-coordination of 5 in 15 could be attributed to a steric repulsion between the monodentate ligands, the trans-coordination of 7c in 16 is at least partly dictated by an electrostatic repulsion between the two cationic ends of the chelate ligand. Due to residual flexibility, the alternative cis-coordination of 7c is indeed not sterically forbidden, as indicated by the X-ray crystal structure of the complex trication [(cis-η2-7c)Rh(MeCN)2]+ involving a harder positively charged Rh(I) center.27 Very recently, the trans-coordination mode of 7c at neutral transition metal centers has been further illustrated and analyzed by the crystal structure and DFT calculations of the (trans-η2-7c)PdCl2 complex.36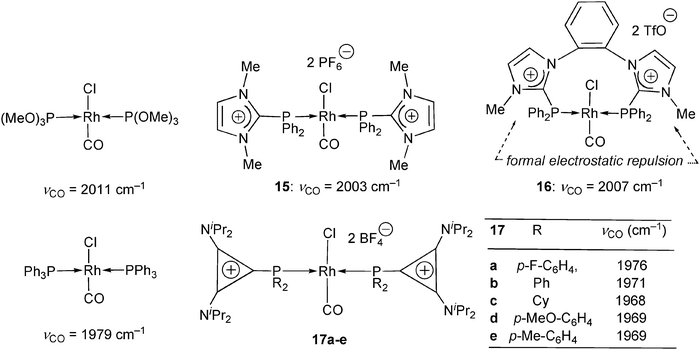 | ||
| Scheme 8 Trans-coordinated rhodiumcarbonyl complexes of trimethylphosphite,35 the monodentate and chelating N-methyl-imidazoliodiphenylphosphine ligands 5 and 7c (Schemes 2 and 3),27,34 and the diaminocyclopropenyliophosphine ligands 12a–e (Scheme 6).30 | ||
The RhCl(CO) complexes of the bis(diisopropylamino)cyclopropenylidene (BAC)-phosphenium ligands 12a–e (Scheme 6) also adopt a trans-coordinative mode (Scheme 8). The corresponding IR stretching frequencies show that these ligands are stronger donors than phosphites and diphenylimidazoliophosphines 5 and 7c, and even slightly stronger than triphenylphosphine (νCO(PPh3) = 1979 cm−1 > νCO(12b) = 1971 cm−1: Scheme 8). It was indeed shown that BAC itself is a stronger donor (νCO[RhCl(CO)2(BAC)] = 2031 cm−1) than related unsaturated NHCs (νCO[RhCl(CO)2(NHC)] ≈ 2041 cm−1).37 The phosphenium character of the PPh2 group of 12b is therefore markedly lowered by the coordination of BAC. This suggests that the “diaminocyclopropenylylidene–phosphenium adducts” 12b and congeners (Scheme 6) could be more accurately referred to as “diaminocyclopropenyliophosphines” (see Scheme 1).
4. Intrinsic reactivity of carbeniophosphines
4.1. Carbeniophosphines as precursors of NHC ligands
As detailed in the introduction (Scheme 1), the dative nature of a covalent bond A–B is determined by an energetic criterion, namely by its preference for a heterolytic dissociation mode A–B → A+ + |B−versus the corresponding homolytic mode A–B → A˙ + ˙B.38 The first experimental evidence of the cleavage of a C+–P bond in a carbeniophosphane was reported in 2004 by Baker et al.,39 who showed that the NHC–diaminophosphenium adduct 18 (stabilized by enhanced delocalization of the positive charge over the phenyltriazolium ring) can dissociate and react with the platinum(0) complex [Pt(PPh3)3] to give the three-coordinate [Pt(phosphane)(phosphenium)(carbene)]+ complex cation 19 (Scheme 9). Although the mechanism was not investigated, the process consists of the displacement of two PPh3 ligands by the strongly σ-donating NHC and the phosphenium moieties resulting from the heterolytic cleavage of 18. The ultimate σ-donating/π-accepting behaviour of the diazaphospholidine unit in 19 is, however, determined by the resonance of the diaminophosphenium form (assigning a formal 0 oxidation state to the Pt atom) with the diaminophosphide form (assigning a formal +2 oxidation state to the Pt atom).![The synthesis of a [Pt(phosphane)(phosphenium/phosphide)(carbene)]+ complex 19 from the NHC–phosphenium precursor 18.39](/image/article/2012/NJ/c1nj20808j/c1nj20808j-s9.gif) | ||
| Scheme 9 The synthesis of a [Pt(phosphane)(phosphenium/phosphide)(carbene)]+ complex 19 from the NHC–phosphenium precursor 18.39 | ||
More nucleophilic than the above Pt(0) complex, are the Cl− and nBu− anions, that were recently shown to induce the heterolytic cleavage of the N2C–P bond of BIMIONAP 6b (Scheme 3) to give chlorodiphenylphosphine and a transient phosphino-NHC that was trapped in situ by either protonation (to give the H-MIOP phosphino-NHC precursor 20),40 sulfurization (to give 21), or hydrolysis (to give the phosphinoformamide 22) (Scheme 10).17
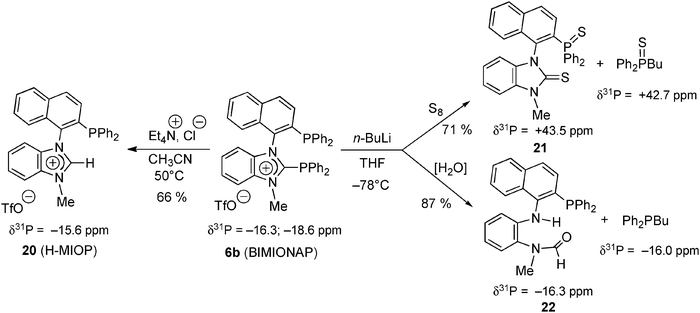 | ||
| Scheme 10 Cleavage of the N2C+–P bond of free BIMIONAP with chloride and alkyl anions, followed by treatment with protic sources (CH3CN, H2O) or elemental sulfur (S8).17 | ||
The same basic reactivity was observed from coordinated BIMIONAP in the palladium complexes 23–24. The NHC moieties released upon cleavage of the N2C+–P bond were instantly and selectively trapped in the coordination sphere of the Pd(II) center to give the NHC-phosphine palladium(II) complexes 25–26 (Scheme 11).
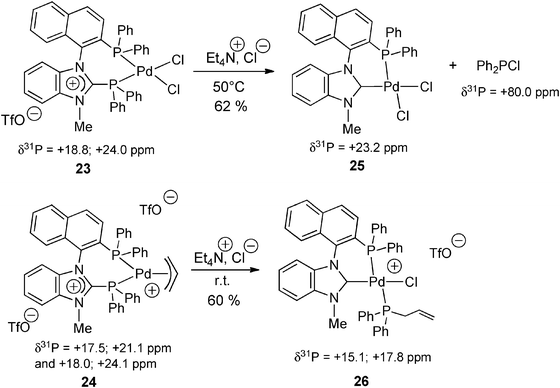 | ||
| Scheme 11 Anion-induced cleavage of the N2C+–P bond of the BIMIONAP ligand within the coordination sphere of Pd(II) centers, and formation of NHC–phosphine palladium complexes 25–26.17 | ||
The above procedures were applied from enantiopure (R)- or (S)-BIMIONAP and PdCl2 complexes thereof (obtained from the resolved enantiomers of BIMINAP 6a: Scheme 3) to give enantiopure (R)- or (S)-H-MIOP (20) and the corresponding NHC–phosphine complexes (R)-25 and (S)-25, respectively.41 To the best of our knowledge, this result constituted the first example of an enantiomerically pure (and atropochiral) amidiniophosphine, thus opening new perspectives in asymmetric catalysis.
Whatever the initial site of the attack by the relatively weak chloride nucleophile, the observed selective cleavage of the N2C+–P bond of BIMIONAP under mild conditions provides strong experimental evidence of its dative nature. The N2C → P+ nature of the bond was also supported by theoretical studies from DFT calculations. These results demonstrate that amidiniophosphines are indeed more accurately described as NHC–phosphenium adducts, thus confirming a previous theoretical suggestion by Macdonald et al.42
4.2. Carbeniophosphines as precursors of highly reactive species
NHCs are used as “stabilizing ligands” for non-metallic electrodeficient species of low coordination number other than phosphenium cations. Such a stabilization was recently described for p-block elements in the zero oxidation state (:Si![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Si:, :GeGe:, ::P–P::, Pn, and C1 fragments),43 germylenes (Ge(II)),44 silylenes (Si(II)),45 and borenium (B(III))46 or tellurium (Te(II))47 cations. In this field, NHC–phosphenium adducts are valuable precursors of other “NHC-stabilized” reactive phosphorus species.
Si:, :GeGe:, ::P–P::, Pn, and C1 fragments),43 germylenes (Ge(II)),44 silylenes (Si(II)),45 and borenium (B(III))46 or tellurium (Te(II))47 cations. In this field, NHC–phosphenium adducts are valuable precursors of other “NHC-stabilized” reactive phosphorus species.
Reaction of NHCs with PCl3 following the route A procedure (Scheme 2) affords NHC → PCl3 adducts 27, which, in the solid state, exhibits a trigonal-bipyramidal configuration of the four-coordinate P atom (with two Cl atoms in the axial positions). One of the P–Clax bonds is, however, significantly longer than the others, thus indicating that 27 is a resonance hybrid of the hypervalent form 27a and the dichlorophosphinoimidazolium chloride all-octet form 27b (Scheme 12).48Reduction of 27 with potassium graphite afforded the diphosphorus complexes 28, which can be considered as bis-phosphinidene units coordinated by two NHC fragments (28c, Scheme 12). A similar adduct was recently isolated by Bertrand et al. by reaction of white phosphorus with a cyclic (alkyl)(amino)carbene (CAAC).49
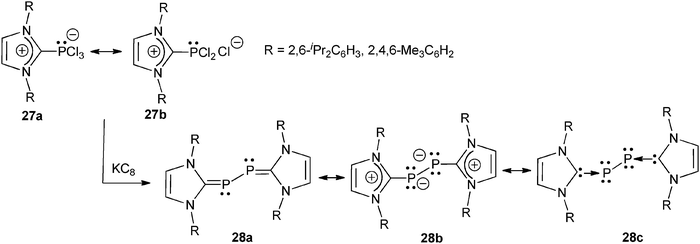 | ||
| Scheme 12 The preparation of NHC-stabilized diphosphorus (P2) from NHC-PCl3 adducts 27, and corresponding resonance forms.48 | ||
As previously mentioned (Section 2.1), the “true” NHC–dichlorophosphenium adduct 5 (where one of the chloride ions of 27 is replaced by a non-coordinating triflate anion) can be quantitatively generated by reaction of PCl3 with the silylimidazolium salt 3 acting as an “onio-substituent transfer” reagent (Scheme 2 route C, and Scheme 13).24 Reaction of PCl3 with two equivalents of 3 afforded the air- and moisture-sensitive P-chlorinated bis(imidazolio)phosphine 29 in excellent yield. The analogous P-phenylated dication 32 was also obtained by Andrieu et al. by reaction of PhPCl2 with 1,3-dimethylimidazolium-carboxylate 2 (Schemes 2 and 13).50 The P-chlorinated adducts 5 and 29 were reported to be valuable precursors of cyano- or azido-P-substituted NHC–phosphenium adducts 30 and 31, respectively.24
![The synthesis of [NHC–PCl2]+ and [(NHC)2–PCl]2+ cations 5 and 29 using the “onio-substituent transfer” reagent 3, and subsequent P-functionalization (see also Scheme 2, route C).24](/image/article/2012/NJ/c1nj20808j/c1nj20808j-s13.gif) | ||
| Scheme 13 The synthesis of [NHC–PCl2]+ and [(NHC)2–PCl]2+ cations 5 and 29 using the “onio-substituent transfer” reagent 3, and subsequent P-functionalization (see also Scheme 2, route C).24 | ||
Switching from diaminocarbenes to CAACs allows for tuning the redox chemistry of the carbene–phosphenium adducts. The CAAC–phosphenium adduct 33, obtained by reaction of the free CAAC51 with (2,2,6,6-tetramethylpiperidino)phosphine dichloride, can thus be selectively reduced to the phosphaalkene 34. The latter can then undergo a one-electron oxidation by treatment with the trityl cation to afford the phosphinyl radical cation 35, which was shown to be very stable both in solution and in the solid state (Scheme 14).52EPR measurements and theoretical calculations of 35 suggested that the unpaired electron is mainly localized at the phosphorus atom with a small spin density at the nitrogen atoms.
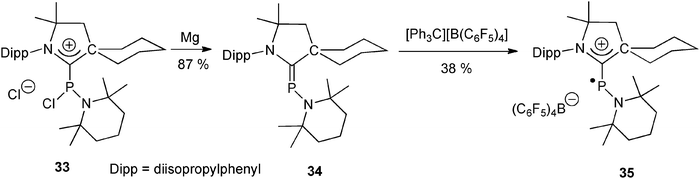 | ||
| Scheme 14 The generation of a stable localized phosphinyl radical 35 from the CAAC–phosphenium adduct 33, through the intermediate formation of phosphaalkene 34.52 | ||
In order to functionalize the C4 position of imidazol-2-ylidene NHCs, and thus tune their donor ability, Bertand et al. described an efficient isomerization procedure from 2-functional imidazoliums. Treatment of the 2-phosphinylimidazolium 36 with a strong base (KHMDS) was thus reported to result in the formation of the 4-phosphinyl phosphinocarbene 38 (Scheme 15). The proposed mechanism involves a deprotonation of 36 to the aNHC (”abnormal NHC”) intermediate 37.53
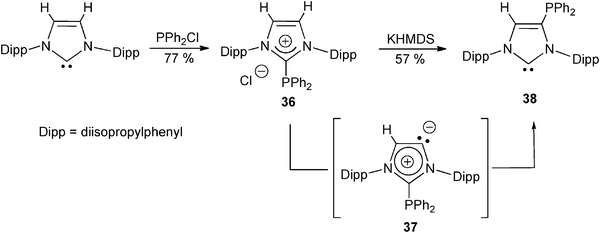 | ||
| Scheme 15 The preparation of a 4-phosphinyl imidazol-2-ylidene 38 from the 2-phophinylimidazolium 36.53 | ||
5. Catalytic properties of carbeniophosphine complexes
As previously mentioned (Section 3), the conjugated positive charge of carbeniophosphanes provides them with a donating character similar to that of phosphites, which should benefit the catalytic processes requiring complexes of electron-deficient ligands. And indeed, even if examples of catalytic applications of carbeniophosphanes are still scarce, these cationic ligands have been already used in C–C coupling reactions, hydrogenation, hydroformylation, hydrosilylation, and cycloisomerisation processes, which are detailed below.5.1. Catalysis in an ionic liquid phase
To date, amidiniophosphines were mainly used in homogeneous polyphasic catalysis, with an ionic liquid phase. To the best of our knowledge, the first report of a catalytic reaction with an amidiniophosphine ligand was described by Knochel et al. in 2000 for the Negishi palladium-catalyzed cross-coupling reaction of aryl- or benzyl-zinc halides with aryl iodides in an ionic liquid/toluene biphasic solvent system.54 Along the same line, Andrieu et al. reported that the use of the cationic phosphine 39 with electron-withdrawing groups at the phosphorus atom provides an efficient catalytic system for the arylation of arylacetylene substrates with aryl bromide electrophiles (Scheme 16).55 Due to the sensitivity of the amidiniophosphine ligand 39, the catalytic activity, however, decreases with recycling. The observation of pyrrolidinone bromide as a side product could explain the deactivation of the catalytic system by nucleophilic displacement of the NHC moiety by the bromide anion, as recently demonstrated for BIMIONAP with the chloride anion (Scheme 10).17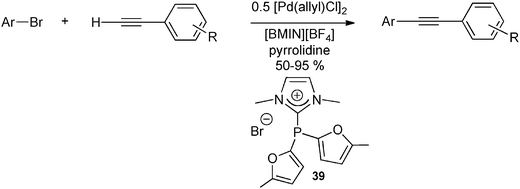 | ||
| Scheme 16 Pd-catalyzed arylation of phenylacetylene derivatives with aryl bromides in presence of the NHC–phosphenium adduct 39 in ionic liquid medium.55 | ||
A multiple recycling of the metal/ligand system was also reported for the platinum-catalyzed hydrogenation of m-chloronitrobenzene in non-acidic media. The authors showed that the use of a platinum(II) chloride complex of the amidiniophosphine 40 increases the life-time of the catalyst as compared to the related neutral complex [trans-PtCl2(PPh3)2] (Scheme 17). This example represents the first case of highly selective catalyst for the hydrogenation of chloronitroarenes immobilized in an ionic liquid phase.55
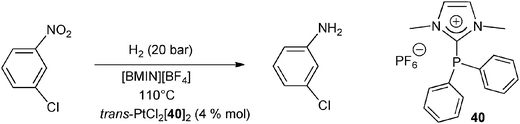 | ||
| Scheme 17 The platinum-catalyzed reduction of m-chloronitrobenzene to m-chloroaniline in presence of the imidazoliophosphine 40 in an ionic liquid medium.55 | ||
Imidazoliophosphine ligands were also successfully applied in biphasic Rh-catalyzed hydroformylation of 1-octene,56Heck reaction of aryl iodides or bromides,57 and hydrosilylation of olefins.58
5.2. Catalysis in a non-ionic phase
Amidiniophosphines were used for the first time in a non-ionic phase for the catalysis of a Sonogashira-type coupling reaction. In particular, it was shown that the BIMIONAP ligand 6b is more efficient than its neutral BIMINAP parent 6a for the reaction of benzoyl chloride with phenylacetylene (Scheme 18).26a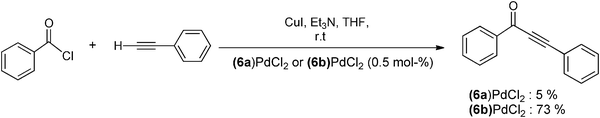 | ||
| Scheme 18 A Sonogashira-type coupling reaction catalyzed by BIMIONAP- and BIMINAP-PdCl2 complexes.26a | ||
The results were analyzed in terms of the difference in electrophilicity of the assumed neutral and cationic intermediates reacting in the limiting step of the catalytic cycle.
More recently, gold complexes of cyclopropenylylidene–phosphenium adducts 12a–e (Scheme 6) were prepared from [(Me2S)AuCl], with subsequent exchange of the chloride anion by the less coordinating bis(trifluoromethanesulfonyl)imidate anion. The corresponding pre-catalysts were tested in the cycloisomerization of enynes, affording good to excellent yields that, in a few cases, surpassed those obtained with Ph3P-derived pre-catalysts. Taking advantage of the cationic nature of the ligands, the authors demonstrated the recycling performance of the catalyst 41 for the cycloisomerzation of an enyne (Scheme 19).30
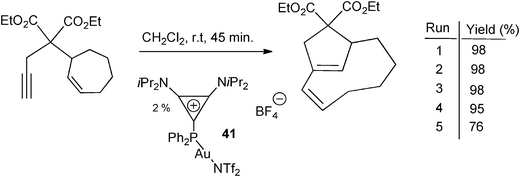 | ||
| Scheme 19 Gold catalyst recycling in the cycloisomerization of an enyne model in the presence of the cationic phosphine 41.30 | ||
6. Conclusion
Although the identification of a novel “class” of phosphorus ligands of transition metals could be considered as a more and more difficult challenge,27 the singular and versatile features surveyed above suggest that carbeniophosphanes deserve to be considered as possible options. Indeed, besides phosphites, they could constitute an alternative for catalytic processes requiring electron-deficient ligands, and some reactions have already been demonstrated to be catalyzed by such cationic ligands. Although their moderate stability (cleavage of the C–P bond under nucleophilic conditions) may be a limitation for their large development, recent reports have shown that modification of the current NHC donor moiety, e.g. by a cyclopropenylidene unit, could allow this problem to be overcome. Alternatively, modification of the substituents at the phosphorus atom (e.g. by amino- or oxy-groups) should allow the tuning of the ‘electronics’ for exploring the stability limit of such systems through the stabilization of the a priori dissociable phosphenium fragment R2P:+. Moreover, due to the availability of the lone pair of the phosphorus atom (as demonstrated in the corresponding complexes), oxidized adducts of carbeniophosphanes (R2C+–PX; X = O, S, Se) are also challenging targets deserving investigation. Finally, carbeniophosphines stand not only as electron-poor phosphorus ligands of transition metals, but also as valuable precursors of stabilized versions of highly reactive species, such as phosphorus radicals or abnormal carbenes.
Acknowledgements
Besides the Ministère de l'Enseignement Supérieur de la Recherche et de la Technologie and the Université Paul Sabatier, the authors thank the Centre National de la Recherche Scientifique and the ANR program (ANR-08-JCJC-0137-01) for financial support.References
- (a) J. F. Young, J. A. Osborn, F. H. Jardine and G. Wilkinson, Chem. Commun., 1965,(7), 131–132 RSC; (b) J. A. Osborn, F. H. Jardine, J. F. Young and G. Wilkinson, J. Chem. Soc. A, 1966, 1711–1732 RSC; (c) D. N. Lawson, J. A. Osborn and G. Wilkinson, J. Chem. Soc. A, 1966, 1733–1736 RSC.
- (a) E. W. Abel, F. G. A. Stone and G. Wilkinson, Comprehensive Organometallic Chemistry II, Pergamon, 1995, vol. 7–9 Search PubMed; (b) E. Jacobsen, A. Pfaltz and H. Yamamoto, Comprehensive Asymmetric Catalysis, Springer, New York, 1999, vol. 1–3 Search PubMed; (c) A. Grabulosa, J. Granell and G. Muller, Coord. Chem. Rev., 2007, 251, 25–90 CrossRef CAS; (d) T. Hayashi, Acc. Chem. Res., 2000, 33, 354–362 CrossRef CAS.
- (a) M. Kruppa and B. Koenig, Chem. Rev., 2006, 106, 3520–3560 CrossRef CAS; (b) K. Muniz, Angew. Chem., Int. Ed., 2005, 44, 6622–6627 CrossRef CAS.
- (a) S. Díez-González, N. Marion and S. P. Nolan, Chem. Rev., 2009, 109, 3612–3676 CrossRef CAS; (b) F. E. Hahn and M. C. Jahnke, Angew. Chem., Int. Ed., 2008, 47, 3122–3172 CrossRef CAS; (c) Y. Canac and R. Chauvin, Eur. J. Inorg. Chem., 2010, 2325–2335 Search PubMed; (d) D. Bourissou, O. Guerret, F. P. Gabbaï and G. Bertrand, Chem. Rev., 2000, 100, 39–91 CrossRef.
- (a) C. A. Fleckenstein and H. Plenio, Chem. Soc. Rev., 2010, 39, 694–711 RSC; (b) R. Martin and S. L. Buchwald, Acc. Chem. Res., 2008, 41, 1461–1473 CrossRef CAS.
- (a) M. Melaimi, M. Soleilhavoup and G. Bertrand, Angew. Chem., Int. Ed., 2010, 49, 8810–8849 CrossRef CAS; (b) J. Vignolle, X. Cattoën and D. Bourissou, Chem. Rev., 2009, 109, 3333–3384 CrossRef CAS; (c) Y. Canac, M. Soleihavoup, S. Conejero and G. Bertrand, J. Organomet. Chem., 2004, 689, 3857–3865 CrossRef CAS.
- (a) Late transition metal complexes of neutral η1-carbon ligands, Top. Organomet. Chem., ed. R. Chauvin and Y. Canac, Springer, 2010, vol. 30, pp. 1–252 Search PubMed; (b) W. C. Kaska and K. A. Ostoja Starzewski, in Ylides and Imines of Phosphorus, ed. A. W. Johnson, John Wiley & Sons, New York, 1993, ch. 14 Search PubMed.
- (a) V. Farina and B. Krishman, J. Am. Chem. Soc., 1991, 113, 9585–9595 CrossRef CAS; (b) N. G. Andersen, R. McDonald and B. A. Keay, Tetrahedron: Asymmetry, 2001, 12, 263–269 CrossRef CAS; (c) M. P. Magee, W. Luo and W. H. Hersh, Organometallics, 2002, 21, 362–372 CrossRef CAS; (d) R. A. Baber, M. L. Clarke, K. M. Heslop, A. C. Marr, A. G. Orpen, P. G. Pringle, A. Ward and D. E. Zambrano-Williams, Dalton Trans., 2005, 1079–1085 RSC.
- (a) G. Celentano, T. Benincori, S. Radaelli, M. Sada and F. Sannicolò, J. Organomet. Chem., 2002, 643, 424–430 Search PubMed; (b) J. J. Becker, L. J. Van Orden, P. S. White and M. R. Gagné, Org. Lett., 2002, 4, 727–730 CrossRef CAS; (c) E. Pizzo, P. Sgarbossa, A. Scarso, R. A. Michelin and G. Strukul, Organometallics, 2006, 25, 3056–3062 CrossRef CAS.
- (a) M. Diéguez and O. Pàmies, Acc. Chem. Res., 2010, 43, 312–322 CrossRef CAS; (b) S. Mano, K. Nozaki and H. Takaya, J. Am. Chem. Soc., 1993, 115, 7033–7034 CrossRef CAS; (c) Y. Yan, Y. Chi and X. Zhang, Tetrahedron: Asymmetry, 2004, 15, 2173–2175 CrossRef CAS.
- (a) F. Berhal, O. Esseiva, C. H. Martin, H. Tone, J. P. Genet, T. Ayad and V. Ratovelomanana-Vidal, Org. Lett., 2011, 13, 2806–2809 CrossRef CAS; (b) C. L. Pollock, G. C. Saunders, E. C. M. S. Smyth and V. I. Sorokin, J. Fluorine Chem., 2008, 129, 142–166 CrossRef CAS; (c) M. L. Clarke, D. Ellis, K. L. Mason, A. G. Orpen, P. G. Pringle, R. L. Wingad, D. A. Zaher and R. T. Baker, Dalton Trans., 2005, 1294–1300 RSC.
- (a) J. J. Brunet, R. Chauvin, G. Commenges, B. Donnadieu and P. Leglaye, Organometallics, 1996, 15, 1752–1754 CrossRef CAS; (b) V. Cadierno, J. Francos and J. Gimeno, Chem.–Eur. J., 2008, 22, 6601–6605 Search PubMed; (c) D. J. M. Snelders, G. v. Koten and R. J. M. Klein Gebbink, Chem.–Eur. J., 2011, 17, 42–57 CrossRef CAS.
- (a) N. Kuhn, M. Göhner and M. Steimann, Z. Anorg. Allg. Chem., 2002, 628, 896–900 Search PubMed; (b) J. Andrieu and M. Azouri, Inorg. Chim. Acta, 2007, 360, 131–135 Search PubMed; (c) J. Andrieu, L. Harmand and M. Picquet, Polyhedron, 2010, 29, 601–605 Search PubMed.
- (a) E. Renaud, R. B. Russel, S. Fortier, S. J. Brown and M. C. Baird, J. Organomet. Chem., 1991, 419, 403–415 CrossRef CAS; (b) P. Leglaye, B. Donnadieu, J. J. Brunet and R. Chauvin, R., Tetrahedron Lett., 1998, 39, 9179–9182 Search PubMed; (c) L. Viau and R. Chauvin, J. Organomet. Chem., 2002, 654, 180–186 Search PubMed.
- R. Chauvin, Eur. J. Inorg. Chem., 2000, 577–591 CrossRef CAS.
- (a) K. H. Shaughnessy, Eur. J. Org. Chem., 2006, 1827–1835 CrossRef CAS; (b) C. Li, Chem. Rev., 2005, 105, 3095–3166 CrossRef CAS.
- I. Abdellah, C. Lepetit, Y. Canac, C. Duhayon and R. Chauvin, Chem.–Eur. J., 2010, 16, 13095–13108 CrossRef CAS.
- If the R2C+ carbenium center is not stabilized by interaction of the empty pz orbital with adjacent n orbitals of nitrogen lone pairs, the interaction is replaced by an overlap with the n orbital of the :PR′2 moiety making the corresponding “phosphoniaalkene” form R2C+PR′2 dominating. Such cations, that can also be designated as “phosphaalkeniums” or “methylenephosphoniums”, have been scarcely exemplified. For early examples, see: A. Igau, A. Baceiredo, H. Grützmacher, H. Pritzkow and G. Bertrand, J. Am. Chem. Soc., 1989, 111, 6853–6854 Search PubMed.
- (a) D. J. M. Snelders, G. van Koten and R. J. M. Klein Gebbink, Chem.–Eur. J., 2011, 17, 42–57 CrossRef CAS; (b) D. J. M. Snelders, M. A. Siegler, L. S. von Chrzanowski, A. L. Spek, G. van Koten and R. J. M. Klein Gebbink, Dalton Trans., 2011, 40, 2588–2600 RSC.
- U. Zoller, Tetrahedron, 1988, 44, 7413–7426 CrossRef CAS.
- (a) N. Kuhn, J. Fahl, D. Bläser and R. Boese, Z. Anorg. Allg. Chem., 1999, 625, 729–734 CrossRef CAS; (b) N. Kuhn, M. Göhner and G. Henkel, Z. Anorg. Allg. Chem., 1999, 625, 1415–1416 Search PubMed.
- M. Azouri, J. Andrieu, M. Picquet, P. Richard, B. Hanquet and I. Tkatchenko, Eur. J. Inorg. Chem., 2007, 4877–4883 CrossRef CAS.
- Y. Canac, R. Chauvin, et al. manuscript in preparation.
- J. J. Weigand, K. O. Feldmann and F. D. Henne, J. Am. Chem. Soc., 2010, 132, 16321–16323 CrossRef CAS.
- A. A. Tolmachev, A. A. Yurchenko, A. S. Merculov, M. G. Semenova, E. V. Zarudnitskii, V. V. Ivanov and A. M. Pinchuk, Heteroat. Chem., 1999, 10, 585–597 CrossRef CAS.
- (a) N. Debono, Y. Canac, C. Duhayon and R. Chauvin, Eur. J. Inorg. Chem., 2008, 2991–2999 CrossRef CAS; (b) I. Abdellah, N. Debono, Y. Canac, L. Vendier and R. Chauvin, Chem. Asian. J., 2010, 5, 1225–1231 Search PubMed.
- Y. Canac, N. Debono, L. Vendier and R. Chauvin, Inorg. Chem., 2009, 48, 5562–5568 CrossRef CAS.
- I. V. Komarov, M. Y. Kornilov, A. A. Tolmachev, A. A. Yurchenko, E. B. Rusanov and A. N. Chernega, Tetrahedron, 1995, 51, 11271–11280 CrossRef CAS.
- H. Jansen, F. B. Läng, J. Chris Slootweg, A. W. Ehlers, M. Lutz, K. Lammerstma and H. Grützmacher, Angew. Chem., Int. Ed., 2010, 49, 5485–5488 CrossRef CAS.
- J. Petuskova, H. Bruns and M. Alcarazo, Angew. Chem., Int. Ed., 2011, 50, 3799–3802 CrossRef CAS.
- (a) N. Burford, T. S. Cameron and P. J. Ragogna, J. Am. Chem. Soc., 2001, 123, 7947–7948 CrossRef CAS; (b) N. Burford, P. J. Ragogna, R. McDonald and M. J. Ferguson, J. Am. Chem. Soc., 2003, 125, 14404–14410 CrossRef CAS.
- C. A. Tolman, Chem. Rev., 1977, 77, 313–348 CrossRef CAS.
- Y. Canac, C. Lepetit, M. Abdalilah, C. Duhayon and R. Chauvin, J. Am. Chem. Soc., 2008, 130, 8406–8413 CrossRef CAS.
- J. Andrieu, M. Azouri and P. Richard, Inorg. Chem. Commun., 2008, 11, 1401–1404 Search PubMed.
- M. L. Wu, M. J. Desmond and R. S. Drago, Inorg. Chem., 1979, 18, 679–686 CrossRef CAS.
- Y. Canac, N. Debono, C. Lepetit, C. Duhayon and R. Chauvin, Inorg. Chem., 2011, 50, 10810–10819 Search PubMed.
- G. Kuchenbeiser, B. Donnadieu and G. Bertrand, J. Organomet. Chem., 2008, 693, 899–904 CrossRef CAS.
- R. S. Mulliken and W. B. Person, Annu. Rev. Phys. Chem., 1962, 13, 107–126 CrossRef CAS.
- N. J. Hardman, M. B. Abrams, M. A. Pribisko, T. M. Gilbert, R. L. Martin, G. J. Kubas and R. T. Baker, Angew. Chem., Int. Ed., 2004, 43, 1955–1958 CrossRef CAS.
- Instead of the expected Ph2PCl co-product, Ph2PP(O)Ph2 was observed, which should result from a hydrolytic dimerization of Ph2PCl, whose exact mechanism under the conditions used has not been investigated.
- I. Abdellah, M. Boggio-Pasqua, Y. Canac, C. Lepetit, C. Duhayon and R. Chauvin, Chem.–Eur. J., 2011, 17, 5110–5115 Search PubMed.
- B. D. Ellis, P. J. Ragogna and C. L. B. Macdonald, Inorg. Chem., 2004, 43, 7857–7867 CrossRef CAS.
- (a) D. Martin, M. Soleilhavoup and G. Bertrand, Chem. Sci., 2011, 2, 389–399 RSC; (b) Y. Wang and G. H. Robinson, Dalton Trans., 2011 10.1039/c1dt11165e , in press.
- (a) P. A. Rupar, V. N. Staroverov, P. J. Ragogna and K. M. Baines, J. Am. Chem. Soc., 2007, 129, 15138–15139 CrossRef CAS; (b) A. J. Ruddy, P. A. Rupar, K. J. Bladek, C. J. Allan, J. C. Avery and K. M. Baines, Organometallics, 2010, 29, 1362–1367 CrossRef CAS.
- (a) R. S. Ghadawal, H. W. Roesky, S. Merkel, J. Henn and D. Stalke, Angew. Chem., Int. Ed., 2009, 48, 5683–5686 CrossRef CAS; (b) A. C. Filippou, O. Chernov and G. Schnakenburg, Angew. Chem., Int. Ed., 2009, 48, 5687–5690 CrossRef CAS.
- T. Matsumoto and F. P. Gabbaï, Organometallics, 2009, 28, 4252–4253 CrossRef CAS.
- J. L. Dutton, H. M. Tuononen and P. J. Ragogna, Angew. Chem., Int. Ed., 2009, 48, 4409–4413 CrossRef CAS.
- (a) Y. Wang, Y. Xie, M. Y. Abraham, R. J. Gilliard, Jr., Pingrong Wei, H. F. Schaeffer III, P. v. R. Schleyer and G. H. Robinson, Organometallics, 2010, 29, 4778–4780 CrossRef CAS; (b) Y. Wang, Y. Xie, P. Wei, R. B. King, H. F. Schaefer III, P. v. R. Schleyer and G. H. Robinson, J. Am. Chem. Soc., 2008, 130, 14970–14971 CrossRef CAS.
- (a) O. Back, G. Kuchenbeiser, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2009, 48, 5530–5533 CrossRef CAS; (b) O. Back, B. Donnadieu, P. Parameswaran, G. Frenking and G. Bertrand, Nat. Chem., 2010, 2, 369–373 CrossRef CAS.
- M. Azouri, J. Andrieu, M. Picquet and H. Cattey, Inorg. Chem., 2009, 48, 1236–1242 CrossRef CAS.
- (a) V. Lavallo, Y. Canac, C. Präsang, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2005, 44, 5705–5709 CrossRef CAS; (b) R. Jazzar, R. D. Dewhurst, J. B. Bourg, B. Donnadieu, Y. Canac and G. Bertrand, Angew. Chem., Int. Ed., 2007, 46, 2899–2902 CrossRef CAS.
- O. Back, M. A. Celik, G. Frenking, M. Melaimi, B. Donnadieu and G. Bertrand, J. Am. Chem. Soc., 2010, 132, 10262–10263 CrossRef CAS.
- D. Mendoza-Espinosa, B. Donnadieu and G. Bertrand, J. Am. Chem. Soc., 2010, 132, 7264–7265 CrossRef CAS.
- J. Sirieix, M. Oβberger, B. Betzemeier and P. Knochel, Synlett, 2000, 11, 1613–1615.
- S. Saleh, E. Fayad, M. Azouri, J. C. Hierso, J. Andrieu and M. Picquet, Adv. Synth. Catal., 2009, 351, 1621–1628 CrossRef CAS.
- D. J. Brauer, K. W. Kottsieper, C. Liek, O. Stelzer, H. Waffenschmidt and P. Wasserscheid, J. Organomet. Chem., 2001, 630, 177–184 CrossRef CAS.
- Q. X. Wan, Y. Liu, Y. Lu, M. Li and H. H. Wu, Catal. Lett., 2008, 121, 331–336 CrossRef CAS.
- J. Li, J. Peng, Y. Bai, G. Zhang, G. Lai and X. Li, J. Organomet. Chem., 2010, 695, 431–436 CrossRef CAS.
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2012 |