Amidines bearing benzofuroxan or benzimidazole 1,3-dioxide core scaffolds as Trypanosoma cruzi-inhibitors: structural basis for their interactions with cruzipain†
Alicia
Merlino
a,
Diego
Benitez
a,
Nuria E.
Campillo
b,
Juan A.
Páez
b,
Luzineide W.
Tinoco
c,
Mercedes
González
*a and
Hugo
Cerecetto
*a
aGrupo de Química Medicinal-Laboratorio de Química Orgánica, Facultad de Ciencias-Facultad de Química, Universidad de la República, Iguá 4225, 11400, Montevideo, Uruguay. E-mail: megonzal@fq.edu.uy; hcerecet@fq.edu.uy; Fax: +598 2 5250749; Tel: +598 2 5258618 (ext. 216)
bInstituto de Química Médica, CSIC, Madrid, Spain
cMultiusers Analysis by NMR Laboratory – LAMAR and Laboratory of Analysis and Development of Enzyme Inhibitors - LADIE, NPPN, Federal University of Rio de Janeiro, Rio de Janeiro, Brazil
First published on 4th November 2011
Abstract
Trypanosoma cruzi, the causative agent of Chagas' disease, affects tens of millions of South Americans. One of the most studied T. cruzi-biomolecules as a target for drug development is cruzipain, an essential cysteine proteinase of this parasite. Some of our recent studies identified amidine containing benzofuroxans as hit compounds for cruzipain inhibition with trypanosomicidal activities. Experimental and theoretical studies inspired us to modify these compounds by maintaining the amidine motif and using benzofuroxan and benzimidazole 1,3-dioxide systems as core scaffolds in order to obtain better cruzipain inhibitors. The new amidines had excellent trypanosomicidal activity, with good selectivity indexes, but without improved cruzipain-inhibitory activities compared with the parent compounds. The interaction of amidines with cruzipain has been investigated through a combined NMR -T1-differences, DOSY, and STD- and molecular docking approaches. Despite the low cruzipain-inhibition ability, our data suggest that these designed compounds have relevant structural features, i.e. aromatic groups and protonated moieties with stabilizing complex ability using stacking and electrostatic interactions, respectively, that bind reversibly to cruzipain.
Introduction
Chagas' disease is the third most common disease in Latin America after malaria and schistosomiasis, and affects at least 15 million people with more than 25 million at risk of infection.1 The infectious agent is the protozoan parasite Trypanosoma cruzi (T. cruzi), which causes symptoms progressing from mild swelling to intestinal disease and ultimately heart failure. The current treatment for Chagas' disease, Nifurtimox, (Nfx, Lampit®) or Benznidazole (Bnz, Rochagan®) is inadequate, since the parasite is often not completely eliminated despite chronic administration, and there are unacceptable side effects.2 Moreover, resistance to these agents has emerged.3 For these reasons the development of safer and more effective drugs against Chagas' disease is urgently needed. The use of multitarget-directed ligands (MTDLs) has emerged as a strategy for the development of new drugs to treat Chagas' disease.4 This approach is based on the combination of two or more pharmacophores into a new chemical entity, also defined as a hybrid-drug, and is only just beginning to be used in drug design for the treatment of Chagas' disease. A recent focus of our attention has been developing MTDLs as anti-T. cruzi agents using free-radical releasing moieties linked to DNA-interacting-,5 sterol-biosynthesis-inhibitor-,6 and cruzipain (CP)-inhibitor- pharmacophores.7Recently, the endoproteinase CP has been described as one of the most promising candidates for vaccine and anti-Chagas drug development.8 This enzyme is the most abundant among the cysteine-, serine-, threonine-, and metallo-proteinases, and it is expressed as a complex mixture of isoforms including the lysosomal, as well as an epimastigote-specific pre-lysosomal organelle called reservosome, the associated to the plasma membrane, and the excreted into the medium. CP digests proteins such as casein, bovine serum albumin and denatured hemoglobin at pH = 3–5 and mainly in the reservosome, and synthetic blocked chromogenic and fluorogenic substrates at pH = 7–9 preferring Arg or Lys at the P1 position, and a hydrophobic or a positively charged residue at P2. The peculiar substrate specificity of CP, somehow intermediate between those of cathepsins L and B, where the enzyme is able to accommodate either a hydrophobic or a positively charged residue at P2, is consistent with the presence of Glu at position 205.9 Glu205 is responsible for the binding of Arg at S2 at physiological pH where Glu is deprotonated, and of a hydrophobic amino acid residues, at lower pH, where Glu is neutral.10
Using the MTDLs strategy, we hybridized benzofuroxan or benzimidazole 1,3-dioxide heterocycles and vinylsulfone or amidine moieties to generate compounds with trypanosomicidal activity involving at least two mechanisms of action – modification of mitochondrial membrane potential,11 and CP inhibition.7 Subsequently we identified compounds 1–3 (Scheme 1a) with good/excellent in vitro trypanosomicidal activity or CP-inhibition ability.12 The lower CP-inhibition ability of compound 1 compares well with known CP-inhibitors, like the sulfone VS4 (Scheme 1b), and this encouraged us to analyze the ligand–CP complexes 3D-structures.13 The sulfones 1 and VS4 share some kind of interactions with CP, i.e. hydrogen-bond interaction with Gln19, Gly66, and His159, and differ in others, i.e. interaction with Trp177 where 1 establishes π-stacking while VS4 establishes hydrogen bonding. However, the most relevant difference evidenced between both complexes was the ability of VS4 to establish intramolecular-π-stacking interactions (Scheme 1c) which contributes to stabilization of the VS4-CP complex.
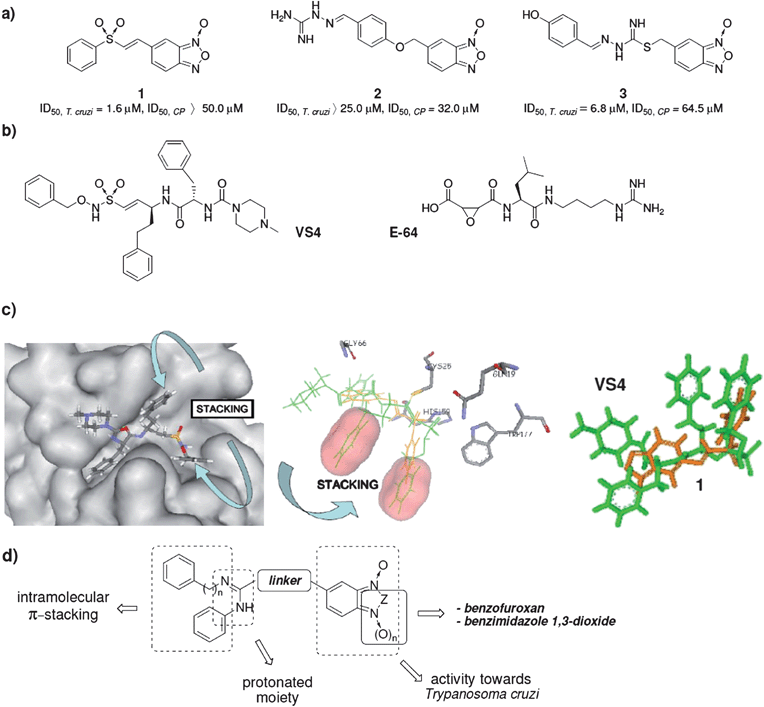 | ||
| Scheme 1 a) Hybrid parent compounds with antitrypanosomal activity and some CP-inhibition capability. b) Well known CP-inhibitors. c) Ligand–CP complex 3D-structures. Left: VS4-CP complex (pdb code 1F2C) showing the intramolecular-π-stacking interaction present in the ligand onto the active site of CP. Middle: Ligand 1 (orange), after the energy optimization, on the CP-active site; inhibitor VS4 (green) is superimposed to show the lack of intramolecular-π-stacking interaction in ligand 1. Right: Ligands 1 (orange) and VS4 (green) are superimposed according to the spatial distribution that they adopt on the CP-active site. d) General structure of the designed amidines. | ||
On the other hand, the amidine moiety present in compounds 2 and 3, that is also present in the well known CP-inhibitor oxyran E-64 (Scheme 1b), is a very interesting motif to be used in CP-inhibitors because it could be protonated in the acidic environment of reservosomes allowing favourable interactions with CP via Glu205, like those that occur with Arg at P2. This idea was recently confirmed by molecular docking studies, where the theoretical results demonstrate that the E-64-amidine group orientates toward Glu205.16
In our efforts to develop selective and novel trypanosomicidal compounds, we designed and synthesized new benzofuroxan and benzimidazole 1,3-dioxides with potential capability to interact with CP through hybridization of the amidine moiety to enable protonation in physiological milieu, and aryl groups able to establish intramolecular-π-stacking (Scheme 1d). The derivatives synthesized were examined for antiproliferative in vitro activity against T. cruzi, Tulahuen 2 strain, and for non-specific cytotoxicity on human red blood cells.
The structural basis for their interactions with the parasite enzyme in solution was mapped by means of NMR experiments, T1-differences, DOSY, and STD. In order to gain further insight into the molecular-level structural features responsible for the experimental observations, molecular docking studies were performed.
Results and discussion
Design and synthesis of new amidines
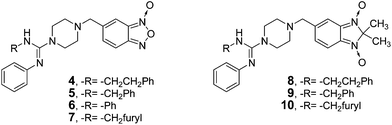
The starting point for the synthesis of the new amidines bearing benzofuroxan or benzimidazole 1,3-dioxide core scaffolds, 4–7 or 8–10, respectively (Table 1, Scheme 2), was the preparation of the corresponding amidine reactant with an adequate nucleophilic-linker which reacts to form 5-bromomethylbenzofuroxan17 (23, Scheme 2). As a first approach the guanylation of thiourea 11 catalyzed by bismuth(III)18 was tried in order to obtain the amidine reactant (I). However, the main reaction was the quinone generation from the phenol moiety, probably due to the Bi(III) oxidation-capability.19 Taking into account that the mechanism of Bi(III) guanylation18 passes through carbodiimide intermediates, carbodiimide 15 (Scheme 2) was treated with p-aminophenol in the absence of bismuth to generate the desired reactant (I). Nevertheless, no reaction occurred after seven days, probably due to the poor nucleophilicity of this aromatic amine. Consequently, the second approach to prepare the amidine reactants involved the reaction of different carbodiimides with a secondary amine, piperazine, which will form part of the linker. Therefore, amidine reactants 19–22 were prepared in moderate yields from phenylisothiocyanate in four steps (see ESI† for complete data related to the syntheses and spectroscopic characterization).20–22 Finally, the desired benzofuroxans, 4–7, were obtained in good to excellent yields by nucleophilic substitution using amidines 19–22 and bromo-derivative 23 (Scheme 2). The benzimidazole 1,3-dioxide derivatives, 8–10, were prepared by treatment of the corresponding benzofuroxan with 2-nitropropane and piperidine as a base in THF at room temperature (Scheme 2). The structures of 4–10 were determined by 1H NMR, 13C NMR, COSY, HSQC, and HMBC experiments, IR and MS. The purity of products was determined by TLC and microanalysis. From the 1H NMR spectra of the amidine reactants and products, some structural features regarding their structural disposition in solution, and at room temperature, could be determined. In acetone-d6 products 4–10 adopted the phenylazamethylidene tautomeric form whereas the reactants 19–22 preferred the R-ylazamethylidene form (as they are represented in Scheme 2) (see ESI, Fig. S1a†). | ||
| Scheme 2 Synthetic procedure used for preparation of amidine derivatives studied herein as cruzipain inhibitors (4–10). | ||
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Compd. | ID50 (μM) | SId | Compd. | ID50 (μM) | SId | ||
| T2a,b | rbcb,c | T2a,b | rbcb,c | ||||
| a T2: Tulahuen 2 strain. b The results are the means of three independent experiments. c rbc: Human red blood cells. d SI: selectivity index (ID50,rbc/ID50,T2). e Problems with compound solubility were observed at higher doses. f From ref. 7c. g ns: not studied. h AmpB: amphotericin B. | |||||||
| 4 | 1.2 ± 0.3 | 37.0 ± 2.2 | 31 | 8 | 1.0 ± 0.1 | >50e | >50 |
| 5 | 7.3 ± 0.5 | 83.0 ± 2.5 | 11 | 9 | 3.0 ± 0.2 | >36e | >12 |
| 6 | 5.8 ± 0.6 | 105 ± 2 | 18 | 10 | 15.0 ± 1.3 | >200e | >13 |
| 7 | 15.0 ± 1.3 | >200e | >13 | ||||
| Nfx | 7.7f | >100e | >13 | ||||
| Bnz | 7.4f | nsg | — | ||||
| AmpB | 0.152 ± 0.006 | 1.5 ± 0.2 | 10 | ||||
CP-inhibition studies
The good anti-T. cruzi activity displayed by derivatives 4–10 (Table 1) encouraged us to investigate the capacity of these amidines to inhibit CP to obtain information on the mechanism of trypanosomicidal activity. The compounds were tested at 25.0, 50.0, and 100.0 μM and at neutral and acidic pH for the ability to inhibit CP. The acidic pH is especially relevant for the protonation of the amidino moiety23 that could change the interaction of the ligand in the active site of the enzyme. The compounds were assayed using Z-phenyl-arginine-7-amido-4-methylcoumarin hydrochloride (Z-Phe-Arg-AMC) as a fluorogenic substrate, and compared with untreated control assays (Table 2). As non-specific compound aggregation could interfere with the assay or promote promiscuous CP inhibition, the experiment was also done in the presence of the non-ionic surfactant Triton X-100 (Table 2) to reduce aggregation and false inhibition.24 In spite of the fact that the new hybrid compounds were more active and selective against T. cruzi than the parent compounds, the CP inhibition capacity could not be improved. In general, the benzimidazole derivatives were better CP inhibitors than the benzofuroxans, with amidine 9 being the best CP inhibitor at pH 7.4. Very interestingly, at pH 5.3 the amidines CP inhibition activities improved in most cases, showing the relevance of protonation for better interactions. At acidic pH, amidine 6 was the best CP inhibitor. Amidine 7 tested in presence of Triton X-100, and at pH 5.3, demonstrated that the effect against CP was not the result of an unrelated inhibitory activity. Additionally, the lack of CP inhibition capability of amidine reactants 19–22 (Table 2), at pH 5.3, confirmed the necessity of special structural requirements for adequate CP-inhibition.| Compd. | % of inhibition at 100.0 μMa | |
|---|---|---|
| pH 7.4 (−T)b | pH 5.3 (−T)/pH 5.3 (+T)c | |
| a The results are the means of three independent experiments. b In buffer, pH 7.4, and absence (−T) of Triton X-100 (see Experimental Section). c In buffer, pH 5.3, and presence (+T) of Triton X-100 (see Experimental Section). d “—”: not studied. e Benzimidazoles were not studied at 5.3 pH due to their stabilities. f mbth: (m-bromophenyl)ethylketone thiosemicarbazone (reference compound). | ||
| 4 | 25.0 ± 2.2 | 34.0 ± 1.3/—d |
| 5 | 29.0 ± 1.6 | 11.0 ± 0.6/— |
| 6 | 12.0 ± 1.4 | 47.0 ± 0.7/— |
| 7 | 0.3 ± 0.2 | 44.0 ± 1.0/45.0 ± 1.2 |
| 8 | 30.0 ± 2.8 | —/— |
| 9 | 34.0 ± 2.0 | —/— |
| 10 | 15.0 ± 1.5 | —/— |
| 19 | — | 3.2 ± 0.5/— |
| 20 | — | 5.4 ± 0.4/— |
| 21 | — | 6.3 ± 1.3/— |
| 22 | — | 10.0 ± 1.1/— |
| mbth | ID50 = 0.10 ± 0.08 μM | —/— |
NMR studies
Measurements of longitudinal relaxation times (T1), diffusion ordered spectroscopy (DOSY), and saturation transfer differences (STD) were used to detect the binding mode and to understand the binding contacts of amidines 4–10 on CP.When a small molecule interacts with a macromolecule, a decrease in T1 is observed due to the slowing of ligand molecular motions as it binds to the biomolecule and the magnitude of this change is related to the intensity of the interaction.25 Therefore, those hydrogens of the small molecule that show a great decrease in their T1 values are the ones that interact more strongly with the macromolecule. This type of information is useful to determine which fragments of the small molecule are involved or not in the molecular interaction and recognition process.26 Additionally, when the small molecule interacts with the macromolecule, changes in its translational movement can be observed. These changes can be evaluated by NMR DOSY experiments measuring the corresponding self-diffusion rates using pulse-field gradient spin-echo sequences.27 The translational self-diffusion rate (D) is a physical parameter which depends on the effective molecular dimensions, the solvent viscosity, and the temperature. Hence, when measured under the same experimental conditions, variations in the D value directly reflect the changes in the effective molecular size. Finally, the STD-experiment involves selectively saturating a resonance that belongs to the macromolecule and when the small molecule binds the saturation will propagate from the selected biomolecule protons being transferred to the binding compound by cross relaxation at the molecule–biomolecule interface.28 When the biomolecule is present in small concentrations, the resulting difference spectrum yields only those resonances that have experienced saturation, namely those of the small molecule that binds to the macromolecule.
Thus, we initially measured the differences in each individual hydrogen T1 and performed the DOSY experiments with 1.3 mM of amidine derivatives 4, 6, and 7, of the amidine reactants 21, and 22, and of parent compound 2, (ratio compound![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CP = 260:1) in deuterated PBS at pH 5.3 and 298 K. Studies with benzimidazoles 8–10 were not performed due to their low stabilities at pH 5.3. In order to maintain the CP-thiol form, DTT was also included in the experiments with amidines 4 and 7. No differences were observed between experiments performed in these conditions and those performed without DTT. In each case, the corresponding proton signals of each compound diffuse at a single rate (D), which rendered average values that are displayed in Table 3 as differences between the absence (Dfree) and the presence of CP (Do). The data suggest that new entities, compound–CP, were produced with higher effective molecular size. In the series of the studied benzofuroxans, 2, 4, 6, and 7, the ΔD values were completely in agreement with the CP inhibition capacity of the compounds at acidic pH, i.e. the parent compound 2, being the best inhibitor of CP suffered the biggest shift in the diffusion coefficient when it interacted with the enzyme. Since D for the ligand–enzyme mixture (Do) is determined according to:
:CP = 260:1) in deuterated PBS at pH 5.3 and 298 K. Studies with benzimidazoles 8–10 were not performed due to their low stabilities at pH 5.3. In order to maintain the CP-thiol form, DTT was also included in the experiments with amidines 4 and 7. No differences were observed between experiments performed in these conditions and those performed without DTT. In each case, the corresponding proton signals of each compound diffuse at a single rate (D), which rendered average values that are displayed in Table 3 as differences between the absence (Dfree) and the presence of CP (Do). The data suggest that new entities, compound–CP, were produced with higher effective molecular size. In the series of the studied benzofuroxans, 2, 4, 6, and 7, the ΔD values were completely in agreement with the CP inhibition capacity of the compounds at acidic pH, i.e. the parent compound 2, being the best inhibitor of CP suffered the biggest shift in the diffusion coefficient when it interacted with the enzyme. Since D for the ligand–enzyme mixture (Do) is determined according to:
| D0 = (Dfree × ffree) + (Dbound × fbound) |
|
|
||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Compd. | ΔDb(cm2s−1)c | ΔT1 (s)d | ID50 CP (μM) | |||||||||||||
| Ha | Hb | Hc | Hd | He | Hf | Hg | Hh | Hl | Hn | Hp | Hq | Hr | Hs | |||
| a The broad characteristics of benzofuroxan proton signals did not allow studies with them in compounds 2, 4, 6, and 7 to be performed. b ΔD = D(compound) − D(compound + CP). c × 10−10. d ΔT1 = T1(compound) − T1(compound + CP). e Protons Hc,d,e, and h were analyzed together. f This value is only indicative because it was not determined. g Protons Hd,e,f,g, and h were analyzed together. h Values in parenthesis are percentages of CP inhibition at 100 μM. | ||||||||||||||||
| 2 | 36 ± 9 | — | — | — | — | — | — | — | — | — | — | 0.09 | 0.22 | 0.15 | 0.41 | 32.0 |
| 4 | 5 ± 1 | 0.08 | 0.10 | 0.13e | 0.14 | 0.01 | 0.11 | 0.08 | 0.06 | — | — | — | >100f | |||
| 6 | 6.7 ± 0.9 | — | — | 0.0 | <0.01g | <0.01 | 0.13 | <0.01 | — | — | — | ∼100f | ||||
| 7 | 10 ± 4 | <0.01 | — | <0.01 | <0.01 | 0.19 | <0.01 | <0.01 | <0.01 | <0.01 | 0.02 | <0.01 | — | — | — | ∼100f |
| 21 | 15 ± 1 | — | — | 0.03 | 0.02g | 0.02 | 0.04 | — | — | — | — | (6.3)h | ||||
| 22 | 18 ± 1 | <0.01 | — | 0.42 | 0.23 | 0.26 | 0.09 | <0.01 | 0.05 | 0.02 | 0.006 | — | — | — | — | (10.0)h |
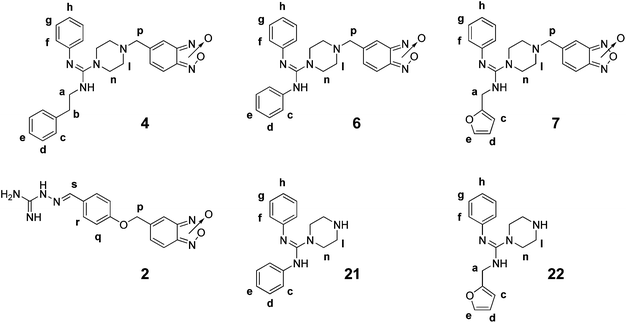
The detailed intermolecular interactions, CP–compounds, were monitored using the differences between the longitudinal relaxation times T1 in both conditions. Clear differences were observed between the best CP-inhibitor, parent compound 2, and the rest of the studied benzofuroxans. The T1 values of protons near to the amidine system in compound 2, Hs, Hr, and Hq, changed more drastically while for the new amidines the strong changes occurred on the other sites, Hf and Hd for 4, Hn for 6, and He for 7 (Table 3). These results indicated that the regions most strongly involved in the interaction with CP are very different. However, when the amidine products, 6 and 7, were compared to the corresponding amidine reactants, 21 and 22, similar regions of interactions were observed, i.e. Hn for 6 and 21 or He for 7 and 22. Consequently, the differences in the CP-inhibition capabilities between these compounds could be the result of the presence of the benzofuroxan system. Unfortunately, it was not possible to study benzofuroxan–protons due to the broad characteristics of these signals as a result of the typical benzofuroxan bond-tautomerism at room temperature.29
STD-NMR experiments provided clear evidence of amidines binding to CP. In all cases, after selective irradiation of the CP resonances, some compound signals appeared in the STD spectra indicating the existence of magnetization transfer from the biomolecule to the inhibitors (Fig. 1). The detailed ligand–CP mapping indicated that binding events occur in specific regions of the studied amidines revealing that methylpiperazinyl moieties (Hp, Hl–m, and Hn–o, Fig. 1) were the main CP-interacting groups. In the case of amidine 4 the phenylethyl moiety (Ha, Hb, Hc, Hd, and He) were also involved in the contact with the CP-backbone (Fig. S2 of the ESI†). The most important magnetized-transfer protons were the methylenes Hl–m from the piperazine moiety. Comparatively, the transference of magnetization from CP to amidine reactants 21 and 22 was much lower than the corresponding magnetization to 6 and 7 (Fig. 1b), entirely in agreement with the CP inhibition capabilities (Table 2).
 | ||
| Fig. 1 STD spectra of the amidines–CP mixture. NS = 256, on-resonance frequency = −0.5 ppm, off-resonance frequency = 20 ppm. a)1H spectra for amidines 6 and 7 with CP (top) STD spectra (bottom). b)1H spectra for the corresponding amidine reactants 21 and 22 with CP (top) and STD spectra (bottom). Note: In these experimental conditions, PBS 5.3 pH at 298 K (for 1H and STD experiments), the amidines (reactants and products) adopted the R-ylazamethylidene tautomers according to chemical shift of protons Hf-f′ (Fig. S1a†). | ||
Molecular docking studies
To get insights into the structural elements responsible for binding to the biological target, and for the eventual design of more selective and potent analogues, 3D models of the complexes between compounds 4–10 and the CP by docking studies were predicted. Molecular docking calculations were performed using FlexiDock, a program that performs docking of conformationally flexible ligands into receptor binding sites and provides control of ligand binding characteristics, taking into account rigid, partially flexible, or fully flexible receptor side chains. FlexiDock incorporates the van der Waals, electrostatic, torsional and constraint energy terms of the Tripos force field, and uses a genetic algorithm (GA) to determine the optimum ligand geometry. GA borrows methodology and terminology from biological (or Darwinian) evolution where an iterative process is used in which the most fit members of a population will have the best chance of propagating themselves in future generations of analysis. Analysis of the enzyme–ligand complex models was based on hydrogen bond, aromatic and hydrophobic interactions predicted with the LPC (Ligand Protein Contact) program and the values of ΔG binding and dissociation constants (Kd) obtained from the docking calculations. In the study parent compound 2 and (m-bromophenyl)ethylketone thiosemicarbazone (mbth) were also included.Interaction energies extracted from the docking study, with amidines 4–6, and 8–10 as monocationic forms (protonated in the amidinyl moiety)30 or with amidines 4–7 as dicationic forms (protonated in both amidinyl and piperazinyl moieties), were in agreement with the experimental results, percentages of CP-inhibition at 7.4 and 5.3 pH, respectively (Table 4). The compounds with the best percentages of CP inhibition at pH 7.4, i.e. benzimidazole 9, or acidic pH, i.e. amidines 6 and 7, displayed the lowest Kd values when modelled as dicationic forms (Table 4). On the other hand, benzofuroxan 5 which displays the worst inhibition capability at pH 5.3 had the highest Kd value. No mathematically valid linear correlation with experimental CP inhibition results was found when the compounds were modelled as monocationic forms. The analysis of docking based on interactions between ligands and CP revealed that compounds 4–10 interacted with CP by hydrophobic interactions with one or more residues present in the substrate-binding cleft. The involved interactions were between the aromatic moieties of amidines and the residues Leu67 and Leu157 (Fig. 2 and Table 5). On the other hand, the amidines established, as proposed (Scheme 1c–d), intramolecular-π-stacking interaction onto the CP-active site contributing to the stabilization of the CP–amidine complexes.
| Compd. | Monocationic forms | PICPc | Dicationic forms | PICPd | ||
|---|---|---|---|---|---|---|
| K d /M | ΔGb/kcal mol−1 | K d /M | ΔGb/kcal mol−1 | |||
| a For parent compound 2 (as monocationic form), Kd = 9.7 × 10−4 M and ΔG = −4.11 kcal mol−1. For mbth (as neutral form), Kd = 6.2 × 10−4M and ΔG = 4.37 kcal mol−1. For monocationic and dicationic form definitions see text. b Values were estimated from the most stable conformers. c PICP: percentage of inhibition at 100.0 μM (Table 2) of CP at 7.4 pH. d PICP: percentage of inhibition at 100.0 μM (Table 2) of CP at 5.3 pH. | ||||||
| 4 | 6.3 × 10−6 | −7.09 | 25 | 8.5 × 10−3 | −2.82 | 34 |
| 5 | 6.6 × 10−6 | −7.06 | 29 | 5.1 × 10−2 | −1.76 | 11 |
| 6 | 1.3 × 10−4 | −5.31 | 12 | 7.2 × 10−3 | −2.92 | 47 |
| 7 | 2.6 × 10−6 | −7.62 | 0.3 | 4.1 × 10−3 | −3.25 | 44 |
| 8 | 1.4 × 10−5 | −6.62 | 30 | 8.9 × 10−3 | −2.79 | — |
| 9 | 9.5 × 10−6 | −6.84 | 34 | 8.1 × 10−3 | −2.85 | — |
| 10 | 4.7 × 10−5 | −5.90 | 15 | 8.4 × 10−3 | −2.83 | — |
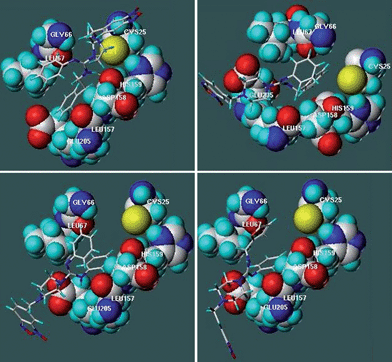 | ||
| Fig. 2 Position of some representative derivatives in the binding-cleft of CP determined by docking. a.CP–4. b. CP–5. c. CP–6. d. CP–7. | ||
| Compd. | d pip-Glu205a | Cys25 (S) | Gly66 (NH) | Gly66 (O) | Leu67 | Leu157 | Asp158 (O) |
|---|---|---|---|---|---|---|---|
| a Mean distance (Å) between piperazinyl moiety (protons l and m) and Glu205 residue. b nc: not correspond. c HB: hydrogen bond. d Bfx: benzofuroxan. e no interaction. f Hph: hydrophobic interactions. g II: ionic interaction. h This ionic interaction is established with piperazinyl group. i Ph: phenyl group. j PhE: phenylethyl group. k Na: amidine nitrogen. l Ben: benzyl group. m Fur: furyl group. n Np: piperazine nitrogen. o Nt: thiosemicarbazone nitrogen. | |||||||
| 2 | ncb | HBc (NBfxd) | HB (NBfx) | —e | Hphf (Bfx) | — | — |
| 4 | — | IIg,h 4.2 Å | — | — | Hph (Phi) | Hph (PhEj) | II (Nak) |
| 5 | 8.6 | — | — | — | — | Hph (Benl) | — |
| 6 | 4.5 | — | — | — | Hph (Ph) | Hph (Ph) | — |
| 7 | 4.8 | — | — | — | Hph (Furm) | Hph (Ph) | — |
| 8 | — | — | — | — | Hph (PhE) | Hph (Ph) | — |
| 9 | — | — | — | — | — | Hph (Ben) | — |
| 10 | — | — | HB (Npn) | — | — | — | — |
| mbth | nc | — | — | HB (Nto) | Hph | — | — |
The theoretical interactions between amidines 4–7 and CP were in accordance with the findings obtained by the experimental NMR data. For example, as reported in Fig. 2a, compound 4 interacts with Leu67 and Leu157 via both phenyl groups in agreement with the T1-differences (Table 3), i.e. according to the docking study, proton Hf is at 3.5 Å of Leu67 residue. Additionally, the piperazinyl group is near to Cys25 (4.2 Å) as T1-differences and STD-experiments also indicate (Table 3, Fig. 1, Fig. S2†). Furthermore, the protonated amidine group of compound 4 establishes ionic interaction (2.8 Å) with Asp158. The FlexiDock analysis located amidines 5–7 in a different manner than amidine 4 (Fig. 2b–d) exposing the benzofuroxan cycle to the solvent. These molecular adjustments positioned the piperazinyl moieties near to Glu205 residue and again the docking results were in accordance with the experimental findings. Compounds 6 and 7, with the highest percentages of CP inhibition at 5.3 pH (Table 2), locate piperazinyl group at 4.5 and 4.8 Å, respectively (Fig. 2), while compound 5, with the poorest capability of CP inhibition at 5.3 pH, situates this moiety at a higher distance to the glutamic acidic residue (8.6 Å, Fig. 2).
Experimental section
Chemistry
:EtOAc, 6:4, and then CH2Cl2:MeOH, 9:1).
5-[4-(N-Phenyl-N′-phenylethyl)amidinopiperazin-1-yl methyl]benzo[1,2-c]1,2,5-oxadiazole N-oxide (4). Brown solid (77%), mp 116.2 °C (d). 1H-NMR (acetone-d6, 400 MHz) δ (ppm): 2.59 (t, 4H, J1 = 4.8 Hz, J2 = 5.2 Hz), 3.06 (t, 2H, J1 = 6.8 Hz, J2 = 7.2 Hz), 3.56 (t, 4H, J = 4.8 Hz), 3.64 (s, 2H), 3.69 (t, 2H, J1 = 6.8 Hz, J2 = 7.2 Hz), 7.14 (dd, 2H, J1 = 8.4 Hz, J2 = 0.8 Hz), 7.19–7.26 (bs, 3H), 7.22 (m, 2H), 7.31 (d, 4H, J = 4.4 Hz), 7.40 (t, 2H, J = 7.6 Hz, J = 0.8 Hz). 13C-NMR (acetone-d6, 100 MHz) δ (ppm): 35.1, 45.8, 48.3, 51.9, 60.9, 121.4, 125.2, 126.6, 128.6, 129.0, 129.6, 138.3, 155.2. ESI MS: m/z = 457 [M+˙ + H+]. IR, ν (cm−1): 3433, 2917, 1622, 1592, 1534, 1487, 1247, 1137, 1000, 853, 751, 699. (Found: C, 68.5; H, 6.16; N, 18.1. C26H28N6O2 requires C, 68.4; H, 6.18; N, 18.4%).
5-[4-(N-Benzyl-N′-phenyl)amidinopiperazin-1-ylmethyl]benzo[1,2-c]1,2,5-oxadiazole N-oxide (5). Beige solid (98%), mp 164.2 °C (d). 1H-NMR (acetone-d6, 400 MHz) δ (ppm): 2.66 (t, 4H, J1 = 4.8 Hz, J2 = 5.2 Hz), 3.64 (s, 2H), 3.70 (t, 4H, J1 = 4.8 Hz, J2 = 5.2 Hz), 4.70 (s, 2H), 7.20 (dd, 2H, J1 = 7.8 Hz, J2 = 2.4 Hz), 7.28–7.50 (bs, 3H), 7.33 (m, 5H), 7.59 (d, 2H, J = 6.8 Hz). 13C-NMR (acetone-d6, 100 MHz) δ (ppm): 47.0, 48.5, 51.9, 60.9, 121.6, 125.3, 127.8, 128.0, 128.6, 129.2, 129.5, 137.8, 155.3. ESI MS: m/z = 443 [M+˙ + H+]. (Found: C, 67.6; H, 5.90; N, 18.8. C25H26N6O2 requires C, 67.9; H, 5.92; N, 19.0%).
5-[4-(N,N′-Diphenyl)amidinopiperazin-1-ylmethyl]benzo[1,2-c]1,2,5-oxadiazole N-oxide (6). Brown oil (42%). 1H-NMR (acetone-d6, 400 MHz) δ (ppm): 2.57 (t, 4H, J1 = 4.8 Hz, J2 = 5.2 Hz), 3.46 (t, 4H, J1 = 4.8 Hz, J2 = 5.2 Hz), 3.64 (s, 2H), 6.92 (t, 2H, J = 7.2 Hz), 7.00 (d, 4H, J = 8.0 Hz), 7.22 (dd, 4H, J1 = 7.2 Hz, J2 = 1.2 Hz), 7.45–7.60 (bs, 3H). 13C-NMR (acetone-d6, 100 MHz) δ (ppm): 46.9, 52.2, 61.4, 120.4, 122.2, 125.6, 128.2, 128.9, 151.0, 154.6. ESI MS: m/z = 429 [M+˙ + H+]. (Found: C, 67.5; H, 5.29; N, 19.2. C24H24N6O2 requires C, 67.3; H, 5.65; N, 19.6%).
5-[4-(N-Furyl-N′-phenyl)amidinopiperazin-1-ylmethyl]benzo[1,2-c]1,2,5-oxadiazole N-oxide (7). Beige solid (52%), mp 152.2–153.0 °C. 1H-NMR (acetone-d6, 400 MHz) δ (ppm): 2.64 (t, 4H, J1 = 4.8 Hz, J2 = 5.2 Hz), 3.63 (t, 4H, J1 = 4.8 Hz, J2 = 5.2 Hz), 3.65 (s, 2H), 4.76 (s, 2H), 6.34 (dd, 1H, J1 = 3.2 Hz, J2 = 1.8 Hz), 7.42–7.57 (bs, 3H), 6.40 (d, 1H, J = 3.2 Hz), 7.18 (m, 1H), 7.25 (m, 2H), 7.38 (m, 2H), 7.51 (dd, 1H, J1 = 1.8 Hz, J2 = 0.8 Hz). 13C-NMR (acetone-d6, 100 MHz) δ (ppm): 40.6, 48.3, 51.9, 60.98, 108.6, 110.5, 121.5, 124.9, 129.0, 129.5, 142.8, 150.1, 155.3. ESI MS: m/z = 433 [M+˙ + H+]. (Found: C, 64.1; H, 5.60; N, 19.2. C23H24N6O3 requires C, 63.9; H, 5.59; N, 19.4%).
:EtOAc, 6:4).
2,2-Dimethyl-5-[4-(N-phenyl-N′-phenylethyl)amidinopiperazin-1-ylmethyl]-2H-benzimidazole 1,3-di-N-oxide (8). Burgundy solid (20%), mp 105.3 °C (d). 1H-NMR (acetone-d6, 400 MHz) δ (ppm): 1.64 (s, 3H), 1.66 (s, 3H), 2.44 (t, 2H, J = 7.2 Hz), 2.62 (bs, 4H), 3.50 (bs, 4H), 3.57 (t, 2H, J = 7.2 Hz), 3.69 (s, 2H), 7.17 (t, 2H, J = 7.6 Hz), 7.23 (d, 1H, J = 7.6 Hz), 7.33 (m, 5H), 7.37 (t, 2H, J = 8.0 Hz), 7.62 (d, 1H, J = 9.2 Hz), 7.83 (s, 1H), 7.89 (d, 1H, J = 9.2 Hz). 13C-NMR (acetone-d6, 100 MHz) δ (ppm): 24.4, 29.0, 47.5, 51.7, 60.7, 60.8, 96.7, 113.6, 115.6, 120.8, 124.4, 126.1, 128.8, 125.1, 134.3, 138.3, 143.2, 160.0. ESI MS: m/z = 441 (M+˙ − 57). (Found: C, 70.0; H, 6.89; N, 16.9. C29H34N6O2 requires C, 69.8; H, 6.87; N, 16.8%).
5-[4-(N-Benzyl-N′-phenyl)amidinopiperazin-1-ylmethyl]-2,2-dimethyl-2H-benzimidazole 1,3-di-N-oxide (9). Bordeaux solid (34%), mp 120.4 °C (d). 1H-NMR (acetone-d6, 400 MHz) δ (ppm): 1.60 (m, 6H), 2.69 (bs, 4H), 3.72 (bs, 4H), 3.74 (s, 2H), 4.71 (s, 2H), 7.20 (t, 2H, J = 7.2 Hz), 7.32 (m, 6H), 7.45 (d, 2H, J = 6.4 Hz), 7.63 (d, 1H, J = 8.4 Hz), 7.85 (s, 1H), 7.91 (d, 1H, J = 9.2 Hz). ESI MS: m/z = 427 (M+˙ − 57). (Found: C, 69.3; H, 6.96; N, 17.1. C28H32N6O2 requires C, 69.4; H, 6.66; N, 17.3%).
5-[4-(N-Furyl-N′-phenyl)amidinopiperazin-1-ylmethyl]-2,2-dimethyl-2H-benzimidazole 1,3-di-N-oxide (10). Bordeaux solid (40%), mp 118.5–119.0 °C. 1H-NMR (CDCl3, 400 MHz) δ (ppm): 1.70 (s, 6H), 2.56 (bs, 4H), 3.46 (bs, 4H), 3.59 (s, 2H), 4.81 (s, 2H), 6.32 (bs, 1H), 6.43 (bs, 1H), 6.88 (d, 1H, J = 8.8 Hz), 7.06 (m, 2H), 7.19 (m, 2H), 7.37 (m, 3H), 7.67 (s, 1H), 7.78 (d, 1H, J = 9.2 Hz). 13C-NMR (CDCl3, 100 MHz) δ (ppm): 24.2, 41.9, 48.0, 51.6, 61.2, 97.2, 109.4, 110.8, 113.9, 114.4, 115.7, 116.3, 120.8, 125.7, 125.5, 129.9, 141.4, 142.6, 149.5, 159.6. ESI MS: m/z = 417 (M+˙ − 57). (Found: C, 65.5; H, 6.36; N, 17.6. C26H30N6O3 requires C, 65.8; H, 6.37; N, 17.7%).
Biology
Non-specific inhibition was evaluated in the presence of Triton X-100. A stock solution 0.02% (v/v) of the detergent was freshly prepared in 100 mM PBS or acetate buffer (pH 7.3 or 5.3, respectively) with EDTA and DTT to achieve the same final concentrations as in the standard assay. 50 μL of this solution was then added to 50 μL of the reaction mixture to obtain 0.01% of Triton X-100 in the final reaction mixture. This concentration of Triton X-100 was found not to interfere with CP activity.
NMR experiments
NMR experiments were recorded on a Varian VNMR500 (1H 498.9 MHz) with a z-axis gradient coil. The experiments were done with a spectral window of 7022 Hz (12.0 to −2.0 ppm) at 298 K. The compounds were dissolved in DMSO-d6 and then diluted (1/70) in deuterated PBS, pH 5.3, at a final dose of 1.3 mM. In each experiment a solution of CP (1.467 mg mL−1 in PBS) was incorporated in the amidine solutions (final volume of 700 μL in deuterated PBS, pH 5.3) to reach a ratio of amidine:CP of 260:1.
Docking studies
The structures of the ligands, as protonated form for the amidinohydrazones and as neutral form for the thio- and semicarbazones, were built with standard bond lengths and angles using the molecular modeling package SYBYL 8.114 and their energies were minimized using the Conjugate Gradiente algorithm with a conjugated gradient of <0.001 kcal mol−1 convergence criterion provided by the MMFF94 force field35 and MMFF94 electrostatic charges. The ligands considered were superimposed onto the inhibitor present in the reference structure (pdb code 1f2936) but without forming any covalent bond with the enzyme. The ligand–receptor complex was subjected to energy minimization using the MMFF94 force field and MMFF94 electrostatic charges and their energies were minimized using the protocol previously indicated with a conjugate gradient of <0.1 kcal mol−1 convergence criterion. These complexes were the input structure for docking studies using the FlexiDock module.37 During the flexible docking analysis, the ligands and a sphere of 6 Å around the corresponding ligand were considered flexible. For each complex three flexible docking analyses were run. The default SYBYL FlexiDock parameters were utilized in all cases, with maximum and minimum iterations (MI) set for each complex according to MI = [No. of rotatable bonds in the protein + No. of rotatable bonds in the ligand + 6 × 1000–500], obtaining a series of model complexes. All conformations obtained with FlexiDock were clustered using a hierarchical cluster analysis taking into account the score and the distances of ligand moieties to key amino acids present in the active site of the enzyme. We chose the conformation with the highest FlexiDock score (better interactions) and refined the minimization energy step using a conjugate gradient of <0.01 kcal mol−1 convergence criterion. Analysis of the refined receptor–ligand complex models was based on hydrogen bond, aromatic and hydrophobic interactions predicted with the LPC (Ligand Protein Contact) program38 and the values of ΔG binding and dissociation constants obtained from the difference accessible surface area method using the STC (Structural Thermodynamics Calculations) program.39Conclusions
Some new anti-Trypanosoma cruzi are amidines bearing benzofuroxan and benzimidazole 1,3-dioxide core designed as CP inhibitors. In the present study the amidines 4–10 have been analysed as potential CP interacting molecules, through different NMR experiments (T1 differences, DOSY, and STD experiments), and experimentally against the enzyme. Molecular modeling studies have also been performed to deepen the structure–activity relationships in order to design more potent and selective analogs. In detail, by NMR experiments we have monitored the binding events revealing that, specially, the piperazinyl moiety of the studied compound interact strongest with CP and, in some cases, the aromatic appendages are involved. The docking results are in agreement with the experimental NMR data. Indeed, the predicted bioactive conformations of tested compounds present the piperazinyl system establishing ionic interactions with Glu205 and the aromatic appendages give further contribution to CP-S2 binding by their van der Waals contacts with Leu67 and Leu157. This investigation has also highlighted that these new amidines fulfil the original design of lateral chains with intramolecular π-stacking to improve the interaction with CP.The presented results can inspire the synthesis of new anti-T. cruzi agents with the capability to act as CP ligands designed for establishing multiple interactions with CP-S2, improving the affinity for this biological target and contributing to sequence selectivity of trypanosomicidal compounds.
Acknowledgements
Financial support from the RIDIMEDCHAG network (CYTED), the MICINN project (TRA2009_0085, 2010–2011), the Collaborative Project UdelaR (Uruguay) – CSIC (Spain) (#2009UY0003) and the REDCLARA-AECID is acknowledged. We thank PEDECIBA-ANII for scholarships to AM and DB, and PEDECIBA for a fellowship to AM. We thank Dr Graciela Mahler for the gift of the mbth reference compound.Notes and references
- http://www.who.int/tdr .
- H. Cerecetto and M. González, Pharmaceuticals, 2010, 3, 810–838 CrossRef CAS.
- Chagas Disease; Special Programme for Research and Training in Tropical Diseases (TDR), World Health Organization: Geneva, http://www.who.int/tdr/diseases/chagas/direction.htm Search PubMed.
- A. Cavalli and M. L. Bolognesi, J. Med. Chem., 2009, 52, 7339–7359 CrossRef CAS.
- (a) L. Otero, M. Vieites, L. Boiani, A. Denicola, C. Rigol, L. Opazo, C. Olea-Azar, J. D. Maya, A. Morello, R. L. Krauth-Siegel, O. E. Piro, E. Castellano, M. González, D. Gambino and H. Cerecetto, J. Med. Chem., 2006, 49, 3322–3331 CrossRef CAS; (b) M. Vieites, L. Otero, D. Santos, J. Toloza, R. Figueroa, E. Norambuena, C. Olea-Azar, G. Aguirre, H. Cerecetto, M. González, A. Morello, J. D. Maya, B. Garat and D. Gambino, J. Inorg. Biochem., 2008, 102, 1033–1043 CrossRef CAS; (c) M. Vieites, L. Otero, D. Santos, C. Olea-Azar, E. Norambuena, G. Aguirre, H. Cerecetto, M. González, U. Kemmerling, A. Morello, J. D. Maya and D. Gambino, J. Inorg. Biochem., 2009, 103, 411–418 CrossRef CAS.
- (a) A. Gerpe, I. Odreman-Nunez, P. Draper, L. Boiani, J. A. Urbina, M. González and H. Cerecetto, Bioorg. Med. Chem., 2008, 16, 569–577 CrossRef CAS; (b) A. Gerpe, G. Álvarez, D. Benítez, L. Boiani, M. Quiroga, P. Hernández, M. Sortino, S. Zacchino, M. González and H. Cerecetto, Bioorg. Med. Chem., 2009, 17, 7500–7509 CrossRef CAS; (c) A. Gerpe, L. Boiani, P. Hernández, M. Sortino, S. Zacchino, M. González and H. Cerecetto, Eur. J. Med. Chem., 2010, 45, 2154–2164 CrossRef CAS.
- (a) W. Porcal, P. Hernández, M. Boiani, G. Aguirre, L. Boiani, A. Chidichimo, J. J. Cazzulo, N. E. Campillo, J. A. Paez, A. Castro, R. L. Krauth-Siegel, C. Davies, M. A. Basombrío, M. González and H. Cerecetto, J. Med. Chem., 2007, 50, 6004–6015 CrossRef CAS; (b) W. Porcal, P. Hernandez, L. Boiani, M. Boiani, A. Ferreira, A. Chidichimo, J. J. Cazzulo, C. Olea-Azar, M. González and H. Cerecetto, Bioorg. Med. Chem., 2008, 16, 6995–7004 CrossRef CAS; (c) A. Merlino, D. Benitez, S. Chavez, J. Da Cunha, P. Hernández, L. W. Tinoco, N. E. Campillo, J. A. Páez, H. Cerecetto and M. González, Med. Chem. Commun., 2010, 1, 216–228 RSC.
- (a) J. J. Cazzulo, Curr. Top. Med. Chem., 2002, 2, 1261–1271 CrossRef CAS; (b) V. G. Duschak and A. S. Couto, Curr. Med. Chem., 2009, 16, 3174–3202 CrossRef CAS; (c) S. I. Cazorla, F. M. Frank, P. D. Becker, M. Arnaiz, G. A. Mirkin, R. S. Corral, C. A. Guzman and E. L. Malchiodi, J. Infect. Dis., 2010, 202, 136–144 CrossRef CAS.
- A. P. C. A. Lima, J. Scharfstein, A. C. Storer and R. Ménard, Mol. Biochem. Parasitol., 1992, 56, 335–338 CrossRef CAS.
- S. A. Gillmor, C. S. Craik and R. J. Fletterick, Protein Sci., 1997, 6, 1603–1611 CrossRef CAS.
- M. Boiani, L. Piacenza, P. Hernández, L. Boiani, H. Cerecetto, M. González and A. Denicola, Biochem. Pharmacol., 2010, 79, 1736–1745 CrossRef CAS.
- CP-inhibition ability for amidine 3 was measured herein.
- For the VS4-CP complex we used the corresponding crystallographic data from: L. S. Brinen, E. Hansell, J. Cheng, W. R. Roush, J. H. McKerrow and R. J. Fletterick, Structure, 2000, 8, 831–840 CrossRef CAS . For sulfone 1 we built the compound with standard bond lengths and angles using the molecular modeling package SYBYL 8.114 and its energy was minimized using the Conjugate Gradient algorithm with a conjugated gradient of <0.001 kcal mol−1 convergent criteria provided by the MMFF94 force field15 and MMFF94 electrostatic charges. The ligand 1 was superimposed onto the inhibitor VS4 present in the reference structure (pdb code 1F2C) bonding covalently to the ligand 1 to Cys25. The complex was subjected to energy minimization using the MMFF94 force field and MMFF94 electrostatic charges and its energy was minimized using the protocol previously indicated with a conjugate gradient of 0.01 kcal mol−1 convergent criteria.
- SYBYL 8.1, Tripos Inc., 1699 South Hanley Rd., St. Louis, MO 63144, USA, 2008 Search PubMed.
- M. Clark, R. D. Cramer III and N. Van Opdenbosch, J. Comput. Chem., 1989, 10, 982–1012 CrossRef CAS.
- S. C. Bourguignon, D. F. B. Cavalcanti, A. M. T. de Souza, H. C. Castro, C. R. Rodrigues, M. G. Albuquerque, D. O. Santos, G. Gomes da Silva, F. C. da Silva, V. F. Ferreira, R. T. de Pinho and C. R. Alves, Exp. Parasitol., 2011, 127, 160–166 CrossRef CAS.
- C. Olea-Azar, C. Rigol, F. Mendizábal, H. Cerecetto, R. Di Maio, M. González, W. Porcal, A. Morello, Y. Repetto and J. D. Maya, Lett. Drug Des. Discovery, 2005, 2, 294–301 CrossRef CAS.
- (a) S. Cunha, B. R. de Lima and A. R. de Souza, Tetrahedron Lett., 2002, 43, 49–52 CrossRef CAS; (b) S. Cunha and M. T. Rodrigues Jr., Tetrahedron Lett., 2006, 47, 6955–6956 CrossRef CAS.
- N. M. Leonard, L. C. Wieland and R. S. Mohan, Tetrahedron, 2002, 58, 8373–8397 CrossRef CAS.
- J. Lee, J. Lee, M. Kang, M. Shin, J.-M. Kim, S.-U. Kang, J.-O. Lim, H.-K. Choi, Y.-G. Suh, H.-G. Park, U. Oh, H.-D. Kim, Y.-H. Park, H.-J. Ha, Y.-H. Kim, A. Toth, Y. Wang, R. Tran, L. V. Pearce, D. J. Lundberg and P. M. Blumberg, J. Med. Chem., 2003, 46, 3116–3126 CrossRef CAS.
- E. A. Hamed, Int. J. Chem. Kinet., 1996, 29, 515–521 CrossRef.
- E. Tassoni, F. Giannessi, T. Bruneti, P. Pessotto, M. Renzulli, M. Travagli, S. Rajamáki, S. Prati, S. Dottori, F. Corelli, W. Cabri, P. Carminati and M. Botta, J. Med. Chem., 2008, 51, 3073–3076 CrossRef CAS.
- B. Sahu, V. Chenna, K. L. Lathrop, S. M. Thomas, G. Zon, K. J. Livak and D. H. Ly, J. Org. Chem., 2009, 74, 1509–1516 CrossRef CAS.
- (a) J. M. Pereira, R. P. Severino, P. C. Vieira, J. B. Fernandes, M. F. G. F. da Silva, A. Zottis, A. D. Andricopulo, G. Oliva and A. G. Corrêa, Bioorg. Med. Chem., 2008, 16, 8889–8895 CrossRef CAS; (b) R. F. Freitas, I. M. Prokopczyk, A. Zottis, G. Oliva, A. D. Andricopulo, M. T. S. Trevisan, W. Vilegas, M. G. V. Silva and C. A. Montanari, Bioorg. Med. Chem., 2009, 17, 2476–2482 CrossRef CAS.
- G. Veglia, M. Delfini, M. R. del Giudice, E. Gaggelli and G. Valensin, J. Magn. Reson., 1998, 130, 281–286 CrossRef CAS.
- Y. F. Cui, G. Y. Bai, C. G. Li, C. H. Ye and M. L. Liu, J. Pharm. Biomed. Anal., 2004, 34, 247–254 CrossRef CAS.
- I. Alfonso, M. Bolte, M. Bru, M. I. Burguete, S. V. Luis and C. Vicent, Org. Biomol. Chem., 2010, 8, 1329–1339 CAS.
- (a) M. Mayer and B. Meyer, Angew. Chem., Int. Ed., 1999, 38, 1784–1788 CrossRef CAS; (b) M. Mayer and B. Meyer, J. Am. Chem. Soc., 2001, 123, 6108–6117 CrossRef CAS.
- R. K. Harris, A. R. Katritzky, S. Oksne, A. S. Bailey and W. G. Paterson, J. Chem. Soc., 1963, 197–203 RSC.
- D. D. Perrin, Dissociation Constants of Organic Bases in Aqueous Solution, Butterworths, London, 1965; Supplement, 1972 Search PubMed.
- (a) M. Boiani, L. Boiani, A. Denicola, S. Torres de Ortiz, E. Serna, N. Vera de Bilbao, L. Sanabria, G. Yaluff, H. Nakayama, A. Rojas de Arias, C. Vega, M. Rolón, A. Gómez-Barrio, H. Cerecetto and M. Gonzalez, J. Med. Chem., 2006, 49, 3215–3224 CrossRef CAS; (b) L. Boiani, C. Davies, C. Arredondo, W. Porcal, A. Merlino, A. Gerpe, M. Boiani, J. P. Pacheco, M. A. Basombrío, H. Cerecetto and M. González, Eur. J. Med. Chem., 2008, 43, 2229–2237 CrossRef CAS; (c) L. Boiani, A. Gerpe, V. J. Arán, S. Torres de Ortiz, E. Serna, N. Vera de Bilbao, L. Sanabria, G. Yaluff, H. Nakayama, A. Rojas de Arias, J. D. Maya, A. Morello, H. Cerecetto and M. González, Eur. J. Med. Chem., 2009, 44, 1034–1040 CrossRef CAS; (d) D. Benitez, M. Cabrera, P. Hernández, L. Boiani, M. L. Lavaggi, R. Di Maio, G. Yaluff, E. Serna, S. Torres, M. E. Ferreira, N. Vera de Bilbao, E. Torres, S. Pérez-Silanes, B. Solano, E. Moreno, I. Aldana, A. López de Ceráin, H. Cerecetto, M. González and A. Monge, J. Med. Chem., 2011, 54, 3624–3636 CrossRef CAS.
- L. Fernandez, M. Calderón, M. Martinelli, M. Strumia, H. Cerecetto, M. González, J. J. Silber and M. Santo, J. Phys. Org. Chem., 2008, 21, 1079–1085 CrossRef CAS; R. Hinojosa Valdez, L. T. Düsman Tonin, T. Ueda-Nakamura, B. P. Dias Filho, J. A. Morgado-Diaz, M. Sarragiotto and C. Vataru Nakamura, Acta Trop., 2009, 110, 7–14 CrossRef.
- R. S. Ferreira, C. Bryant, K. K. H. Ang, J. H. McKerrow, B. K. Shoichet and A. R. Renslo, J. Med. Chem., 2009, 52, 5005–5008 CrossRef CAS.
- R. Freeman, H. D. W. Hill, B. L. Tomlinson and L. D. Hall, J. Chem. Phys., 1974, 61, 4466–4473 CrossRef CAS.
- M. Clark, R. D. Cramer III and N. Van Opdenbosch, J. Comput. Chem., 1989, 10, 982–1012 CrossRef CAS.
- L. S. Brinen, E. Hansell, J. Cheng, W. R. Roush, J. H. McKerrow and R. J. Fletterick, Structure, 2000, 8, 831–840 CrossRef CAS.
- R. Judson, Genetic algorithms and their use in chemistry, in: K. B. Lipkowitz, D. B. Boyd (ed.), Reviews in Computational Chemistry, vol. 10, VCH Publishers, NewYork, 1997, pp. 1–73 Search PubMed.
- V. Sobolev, A. Sorokine, J. E. Prilusky, E. Abola and M. Edelman, Bioinformatics, 1999, 15, 327–332 CrossRef CAS.
- P. Lavigne, J. R. Bagu, R. Boyko, L. Willard, C. F. B. Holmes and B. D. Sykes, Protein Sci., 2008, 9, 252–264 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Discussion about tautomeric-forms of amidines, Fig. 1S and 2S, and detailed experimental procedures and spectroscopic characterization of amidines reactants 19–22. See DOI: 10.1039/c1md00223f |
| This journal is © The Royal Society of Chemistry 2012 |