Kinetic measurements give new insights into lipid membrane permeabilization by α-synuclein oligomers†‡
Martin
Stöckl
*a,
Mireille M. A. E.
Claessens
a and
Vinod
Subramaniam
*ab
aNanobiophysics, MESA+ Institute for Nanotechnology, University of Twente, P.O. Box 217, 7500 AE Enschede, The Netherlands. E-mail: m.t.stockl@utwente.nl; v.subramaniam@utwente.nl
bNanobiophysics, MIRA Institute for Biomedical Technology and Technical Medicine, University of Twente, P.O. Box 217, 7500 AE Enschede, The Netherlands
First published on 18th October 2011
Abstract
Interactions of oligomeric aggregates of the intrinsically disordered protein α-synuclein with lipid membranes appear to play an important role in the development of Parkinson's disease. The permeabilization of cellular membranes by oligomers has been proposed to result in neuronal death. The detailed mechanisms by which α-synuclein oligomers permeabilize lipid bilayers remain unknown. Two different mechanisms are conceivable. Oligomers may either insert into membranes forming pores through which small molecules can cross the membrane or their interaction with the membrane may disorder the lipid packing, giving rise to membrane defects. Here we show, using kinetic leakage measurements, that α-synuclein oligomer induced impairment of membrane integrity is not limited to the formation of permanent membrane spanning pores. Fast membrane permeabilization could be observed in a fraction of the large unilamellar vesicles. We have also observed, for the first time, that α-synuclein oligomers cause an enhanced lipid flip-flop. In neuronal cells, most of the α-synuclein is not expected to be present in an oligomeric form, but as monomers. In our in vitro experiments, we find that membrane bound monomeric α-synuclein can only delay the onset of oligomer-induced membrane permeabilization, implying that α-synuclein monomers cannot counteract oligomer toxicity.
Introduction
An increasing fraction of the population is affected by neurodegenerative diseases such as Alzheimer's disease and Parkinson's disease. Parkinson's disease (PD) currently occurs in more than 1% of the population at an age of 65 and older.1 Although the death of dopaminergic neurons in the substantia nigra has been identified as the principal cause for the neurological deficits observed in PD,2–4 the pathological events leading to the profound loss of these neurons remain to be elucidated. One of the pathological hallmarks of PD is the appearance of large intracellular protein aggregates or Lewy bodies.5,6 These aggregates are rich in amyloid fibrils formed by the protein α-synuclein. In addition, mutated forms of α-synuclein are associated with hereditary forms of PD.7–9 These observations suggest that α-synuclein is one of the key players in disease progression.α-Synuclein is a 14.5 kDa protein present at high concentrations in the cytoplasm of neurons, and has long been thought to be an intrinsically disordered protein. A very recent report suggests that physiological α-synuclein is predominantly present as a helically-folded tetramer.10 In any case, the intrinsically disordered nature of the recombinantly produced proteinin vitro appears to confer upon it a remarkable conformational plasticity. Circular dichroism spectroscopy of the monomeric protein reveals a spectrum typical for a random coil structure. However, the measured hydrodynamic radius deviates from that calculated for a pure random coil protein, which suggests the existence of residual structure.11–14 Indeed, some studies report long-range electrostatic interactions with other segments of the protein, which may inhibit the aggregation of the protein.14–19 In this view, modifications which change the degree of residual structure may have an impact on the propensity of α-synuclein for aggregation.20 The N-terminal part of the protein contains hexameric repeats that share sequence similarities with the apolipoproteins. Like apolipoproteins, α-synuclein is able to bind to phospholipid bilayers. Upon binding, the N-terminal part of the protein folds into an α-helical structure.21–23Membrane binding is likely to mediate the enrichment of α-synuclein at presynaptic terminals. At the terminals α-synuclein is thought to regulate the stabilization and release of synaptic vesicles.24,25
Although PD risk factors have been extensively evaluated, it is yet unknown which events cause the aggregation of functional α-synuclein into oligomers and fibrils, thus triggering the onset of PD.26 Additionally, the nature of the neurotoxic α-synuclein species is yet to be identified.27,28 There are indications that monomeric and mature fibrillar α-synuclein are benign, while oligomeric forms of the protein seem to be the main toxic species.29–32 The toxic mechanism of the oligomeric α-synuclein species is still unknown. Misfolded aggregated proteins possibly block protein degradation pathways, or inhibit processes associated with autophagy, mitochondria or cellular trafficking.33–35 In addition, α-synuclein oligomers have been shown to permeabilize lipid membranes both in cellular and membrane model systems.28,36–40 The integrity of cellular compartments is strictly regulated and a prerequisite for the survival of cells. Therefore, membrane permeabilization is a feasible mechanism by which α-synuclein oligomers could lead to neuronal death.
In this paper we investigate the impact of α-synuclein oligomers on phospholipid membranes in a model large unilamellar vesicle (LUV) system containing 1-palmitoyl-2-{6-[(7-nitro-2-1,3-benzoxadiazol-4-yl)amino]hexanoyl}-phosphatidylcholine (C6-NBD-PC). Addition of dithionite to the vesicle solution effectively bleaches all accessible fluorophores on the exposed outer leaflet, while C6-NBD-PC in the inner leaflet is protected from bleaching. Membrane permeabilization by α-synuclein oligomers allows an influx of the reductant into the LUVs and complete bleaching of C6-NBD-PC. Analysis of the dithionite influx kinetics revealed a biexponential behavior. We attribute a fast loss of fluorescence to the rapid influx of dithionite into a fraction of the LUVs, and the further slow loss of fluorescence to an enhanced lipid flip-flop that exposes lipids originating from the inner leaflet of unpermeabilized vesicles to the dithionite. We also studied the impact of the fraction of charged lipids and the presence of monomeric α-synuclein on the impairment of the membrane integrity by oligomeric α-synuclein.
Results
Measuring membrane permeabilization by bleaching of C6-NBD-PC
A range of experimental approaches has established that α-synuclein oligomers can permeabilize phospholipid membranes. However, it is still unclear by which mechanism the membrane integrity is impaired. On the one hand α-synuclein oligomers may integrate in the membrane and form pores through which small molecules permeate. On the other hand it is conceivable that the interactions between the α-synuclein oligomers and the phospholipid bilayer destabilize the packing of the lipids and therefore impair the bilayer integrity. A thorough characterization of the leakage kinetics after the addition of the oligomers to lipid bilayers is essential to get an insight into the membrane disruption mechanism.To follow the membrane leakage as a function of the oligomer concentration, 1-palmitoyl-2-oleoyl-phosphatidylglycerol (POPG) LUVs containing 1 mol% of C6-NBD-PC were mixed with α-synuclein oligomers. Bleaching of C6-NBD-PC was initiated by a subsequent addition of 10 mM dithionite (Fig. 1).
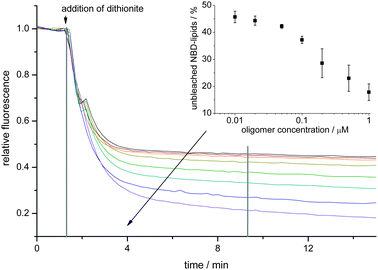 | ||
| Fig. 1 α-Synuclein oligomers cause an influx of dithionite into POPG LUVs in a concentration dependent fashion. To LUVs different concentrations of oligomers were mixed and at the indicated time point 10 mM dithionite was added. Curves for oligomer concentrations of 0, 0.01, 0.02, 0.05, 0.1, 0.2, 0.5 and 1.0 μM (along the direction of the arrow) are shown. After 8 min (vertical line) the remaining fluorescence was measured (see inset). Averages of three independent measurements are shown. Error bars denote the standard deviation. | ||
Immediately after the addition of the dithionite a fast reduction of the C6-NBD-PC fluorescence signal was observed for all oligomer concentrations. After a few minutes the decrease in fluorescence slowed down in an oligomer concentration dependent fashion. In the absence of α-synuclein oligomers the fluorescence signal after dithionite addition rapidly decreased to about 50% of the initial value, which is attributed to complete bleaching of the dithionite-accessible C6-NBD-PC in the outer leaflet of the LUV membrane. Later on, only a slow loss of fluorescence presumably due to photobleaching or to a slow basal permeation of dithionite into the LUVs could be observed. In the presence of α-synuclein oligomers the amount of rapidly bleached NBD-lipids increased with the oligomer concentration (see inset in Fig. 1).
Impact of lipid composition on membrane permeabilization
Previous binding studies using giant unilamellar vesicles showed that the amount of membrane bound α-synuclein was proportional to the fraction of negatively charged lipids present.41 Thus, negatively charged lipids serve as the sole binding sites for α-synuclein monomers. To test whether the fraction of negatively charged lipids has an impact on the ability of oligomers to permeabilize phospholipid bilayers, LUVs were prepared from POPG/1-palmitoyl-2-oleoyl-phosphatidylcholine (POPC) mixtures containing 1 mol% of C6-NBD-PC. After initial bleaching of the NBD-labeled lipids in the outer leaflet, 0.1 μM of α-synuclein oligomers were added. In Fig. 2 the fluorescence time traces for LUV preparations with different surface charge densities are shown.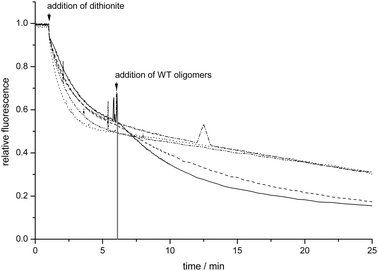 | ||
| Fig. 2 Kinetics of C6-NBD-PC bleaching in LUVs containing different fractions of negatively charged lipids (POPG only: solid, POPG/POPC: 90/10 dashed, 70/30 dash dot, 50/50 dash dot dot, 25/75 dotted). At the indicated timepoint 10 mM dithionite was mixed with the LUVs. 5 min after the dithionite addition, 0.1 μM of α-synuclein oligomers were added. | ||
For POPG/POPC LUVs with a high POPG content the exponential decrease in fluorescence intensity in time is followed by an additional decrease in fluorescence upon addition of α-synuclein oligomers. When the POPG content in the LUVs is lower than 90% no further decrease in fluorescence was observed after the addition of α-synuclein oligomers. This observation indicates that α-synuclein oligomers can only permeabilize LUVs containing high fractions of POPG. Thus, a very high fraction of negatively charged lipids in the membrane apparently makes the phospholipid bilayer susceptible to oligomer-induced leakage. Yet, such high fractions of negatively charged lipids normally are not present in cellular membranes. Since the distribution of the lipids in the membrane is not necessarily homogeneous, a protein-mediated localized enrichment of negatively charged lipids is conceivable, rendering these negatively-charged membrane patches susceptible to permeabilization by α-synuclein oligomers.
Influence of α-synuclein monomers on membrane permeabilization
In neuronal cells most of the α-synuclein is not expected to be present in the oligomeric form, but as monomers. The presence of membrane associated monomeric α-synuclein may thus have a significant impact on the interaction between oligomers and membranes. We therefore characterized the effect of the presence of monomeric α-synuclein on the propensity of oligomers to permeabilize membranes. To this end LUVs (POPG/POPC = 90/10) containing 1 mol% of C6-NBD-PC were mixed with different concentrations of monomers. After bleaching the NBD-labeled lipids in the outer leaflet by dithionite, 0.1 μM of α-synuclein oligomers were added and the kinetics of the NBD fluorescence intensity decrease were recorded (Fig. 3).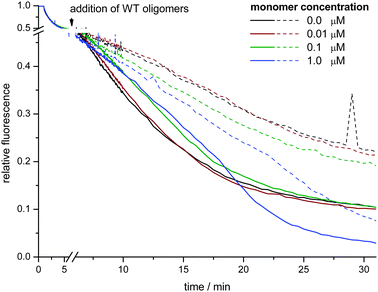 | ||
| Fig. 3 Kinetics of C6-NBD-PC bleaching in LUVs (POPG/POPC = 90/10) in the presence of different amounts of monomeric α-synuclein. The monomers were premixed with the LUVs. C6-NBD-PC in the outer leaflet was bleached by addition of 10 mM dithionite. 5 min after the dithionite addition, 0.1 μM of α-synuclein oligomers were added (full lines). For comparison, influx kinetics curves in the absence of oligomers are also shown (dashed lines). | ||
In the absence of oligomers, no fast additional decrease of fluorescence can be observed, which confirms earlier results that α-synuclein monomers do not readily permeabilize phospholipid bilayers. Merely the presence of a high monomer concentration (1.0 μM) gradually causes an enhanced loss of fluorescence at later timepoints. The fast influx of dithionite caused by the oligomer membrane interactions was delayed in the presence of 0.1 or 1.0 μM monomers. Interestingly, a monomer concentration of 0.1 μM would correspond to a protein/lipid ratio of 1/100 for which a saturation of membrane binding was observed with 1-palmitoyl-2-oleoyl-phosphatidylserine LUVs.42 However, eventually monomers cannot protect the membrane against oligomer-induced permeabilization and the LUVs exhibit the same level of permeabilization as in the absence of the monomers.
Dithionite influx kinetics after membrane permeabilization
In order to characterize the processes involved in the α-synuclein oligomer mediated membrane permeabilization and dithionite influx, POPG LUVs containing 1 mol% C6-NBD-PC were incubated with different concentrations of α-synuclein oligomers. The oligomers were added after initial dithionite bleaching of C6-NBD-PC embedded in the outer leaflet. Subsequently, the kinetics of the loss of fluorescence was measured. The fluorescence data could be well fitted by biexponential decay functions (Fig. S1, ESI‡). Parameters recovered from the fits are plotted in Fig. 4A and B.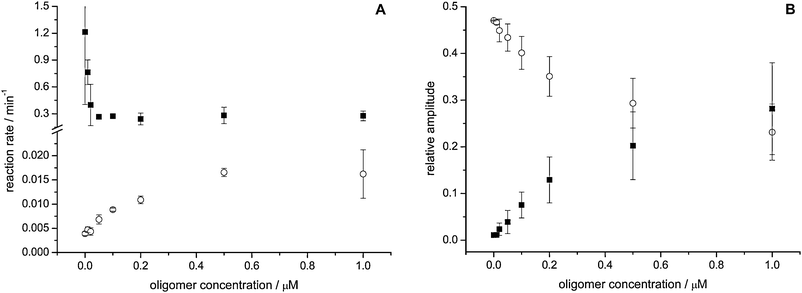 | ||
| Fig. 4 Influx of dithionite into POPG LUVs caused by α-synuclein oligomers is a two phase process. In POPG LUVs C6-NBD-PC was bleached in the outer leaflet by addition of dithionite. After 5 min α-synuclein oligomers were added and the ongoing influx of dithionite was followed. The figures show the reaction kinetics (A) and relative amplitudes normalized to the total fluorescence (B) of the fast (full squares) and the slow (open circles) process. Averages of two independent measurements are shown. Error bars denote the standard deviation. | ||
After oligomer-induced permeabilization, a fast loss of fluorescence can be observed with a rate constant of about 0.3 min−1 (Fig. 4A). The amplitude of this process increases in an oligomer concentration dependent fashion, but the rate constant remains unchanged (Fig. 4B). This observation suggests that the rate constant of the observed fluorescence intensity decay is merely determined by the reaction kinetics of the bleaching reaction. Additionally, a much slower loss of fluorescence is observable. This process is very slow in the absence of α-synuclein oligomers with a rate constant of about 0.003 min−1, which increases in an oligomer concentration dependent fashion to ∼0.02 min−1 at the highest oligomer concentrations (Fig. 4A). These data suggest that two different processes are responsible for the loss of fluorescence. A fast influx of dithionite, occurring in a fraction of the LUVs, shows a rate constant that is independent of oligomer concentration, and is accompanied by a slower process with a concentration dependent rate constant, affecting LUVs that are not permeabilized. With higher α-synuclein oligomer concentration, the relative amplitudes shift in favor of the fast influx.
To test whether the slow rate constant is caused by the exposure of C6-NBD-PC molecules to dithionite due to lipid flip-flop, we labeled POPG LUVs exclusively on the outer leaflet with 1 mol% C6-NBD-PC of total lipids and incubated these vesicles for 60 min with α-synuclein oligomers. C6-NBD-PC in the outer leaflet was bleached by addition of dithionite and the remaining fluorescence signal from C6-NBD-PC that had flipped to the inner leaflet was determined (Fig. 5). This assay clearly shows that α-synuclein oligomers induce lipid flip-flop in the vesicles. For high α-synuclein concentrations a plateau is reached so that apparently at most 10% of the available C6-NBD-PC gets protected in the inner leaflet within 60 min. At a first glance the extent of lipid flip-flop seems to be too low to explain the observed slow process of fluorescence loss. However, a persistent permeability of the vesicles after preincubation with α-synuclein oligomers would expose also the flipped lipids on the inner leaflet to the dithionite, diminishing the observed fluorescence signal. Therefore, we measured the permeability of the vesicle membrane to dithionite for LUVs preincubated for 60 min with oligomers in order to determine the extent of influx of dithionite (Fig. 5).
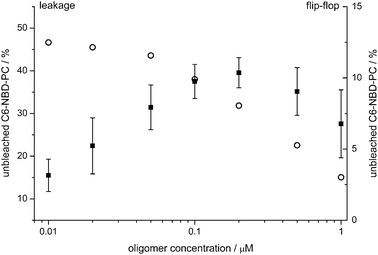 | ||
| Fig. 5 α-Synuclein oligomers cause an enhanced flip-flop of C6-NBD-PC in POPG LUVs in a concentration dependent fashion. POPG LUVs labeled asymmetrically on the outer leaflet (flip-flop, full squares) or symmetrically (leakage, open circles) with 1 mol% C6-NBD-PC were incubated with α-synuclein oligomers for 60 min. 10 mM dithionite was added to bleach all accessible fluorophores. The remaining fluorescence signal relative to the original intensity is shown. While for the leakage assay a single measurement is shown, data points of the flip-flop assay are averages of three independent measurements. Error bars denote the standard deviation. | ||
The leakage assay clearly shows that for α-synuclein oligomer concentrations at which the amount of measured flip-flop levels off, substantial influx of dithionite can be observed. As the fraction of LUVs which becomes permeable to dithionite due to the addition of oligomers does not contribute to the fluorescence signal, the overall amount of lipid flip-flop is strongly underestimated. These data suggest that the enhanced lipid-flip-flop observed indeed may explain the further loss of fluorescence signal.
Discussion
Using a calcein release assay, it has been already well established that α-synuclein oligomers are able to permeabilize phospholipid bilayers.38,40 However, the mechanisms by which these oligomeric protein aggregates impair membrane integrity remain elusive. Using an independent approach, based on the bleaching of C6-NBD-PC by dithionite, we studied factors which render a lipid bilayer susceptible for permeabilization and measured the kinetics of the influx of small molecules into lipid vesicles. As the properties of the α-synuclein oligomers present in the neurons are not known, a variety of protocols is used to trigger α-synuclein aggregation. We have used a protocol that relies on a high protein concentration to trigger the aggregation,39,43 which yields small spherical aggregates, similar to oligomers reported by others.44,45Validation of the dithionite assay
In contrast to other assays measuring the influx or efflux of a reporter, we make use of a fluorescent probe embedded in the phospholipid bilayer, which greatly facilitates the preparation of vesicles by eliminating an additional purification step to separate free dye from dye encapsulated in the LUVs. As the lipid bilayer is normally not permeable to the charged dithionite anion, a specific bleaching of the C6-NBD-PC molecules in the outer leaflet can be achieved.46 Conditions that render the membrane permeable to small molecules allow for access of the dithionite into the vesicle interior, therefore causing fluorescent probes on the inner leaflet to be accessible to the reductant. The measured concentration dependent membrane permeabilization (Fig. 1) agrees very well with data previously measured with a calcein release assay,40 showing that membrane permeabilization is not sensitive to the properties of the small molecules used.Membrane permeabilization by α-synuclein oligomers requires a high fraction of negatively charged lipids
Many reports have shown that in well-ordered lipid bilayers the presence of negatively charged lipids is a prerequisite for interaction of both monomeric and oligomeric α-synuclein.41–43,47–49 The negatively charged lipid headgroups act as binding sites for the positively charged lysine residues in the N-terminal segment of α-synuclein. This interaction, in the case of the monomers, triggers the structural transition from an intrinsically disordered to an α-helical structure. Interestingly, in the oligomers exactly the same regions seem to be involved in membrane binding.50 It is therefore reasonable to assume that the available binding sites, that is, the negatively charged head-groups, determine the amount of α-synuclein oligomers bound at the phospholipid bilayer. Therefore, a linear relation between the amount of membrane-bound oligomeric α-synuclein and the fraction of negatively charged lipids in the vesicle is expected,43 as has been observed for monomeric α-synuclein.41 Moreover, if α-synuclein oligomer binding creates a membrane pore through which small molecules can permeate the lipid bilayer, as is the case for many membrane-spanning proteins, a linear relation should also be observable for the amount of membrane permeabilization by α-synuclein oligomers. However, this is not the case, as the fraction of negatively charged lipids exhibits a sharp cutoff between 70 and 90 mol% of POPG in POPG/POPC LUVs where membrane permeabilization is completely abolished, although membrane bound α-synuclein oligomers are still present. Since a larger number of membrane binding sites would result in a higher local concentration, it is conceivable, for the case that several oligomers bind cooperatively to form a membrane pore, that a nonlinear relation exists between oligomer concentration and membrane permeabilization. However, one would then still expect a more gradual increase in membrane permeabilization than the sharp threshold observed. Additionally, if the α-synuclein oligomers would integrate in a transbilayer fashion, forming a hydrophilic pore, it is reasonable to assume that after binding to the bilayer the insertion into the membrane, mainly driven by short range van der Waals interactions, should readily occur. Thus, this mechanism is not consistent with the observed behavior. Alternatively, if the α-synuclein oligomers permeabilize lipid bilayers by an impairment of the lipid order, creating short-lived membrane defects, the susceptibility of the bilayer to permeabilization would correlate with the degree of lipid order present. In this view, a lipid bilayer containing a high fraction of equally charged lipids would, due to mutual headgroup repulsion, be less stable than a bilayer composed of mainly zwitterionic lipids. Thus, although a moderate fraction of negatively charged lipids yields binding sites for the α-synuclein oligomers,43 the lipids remain sufficiently well-ordered to resist the formation of membrane defects caused by the embedding of α-synuclein oligomers. This model also agrees very well with the findings that the susceptibility of LUVs to membrane permeabilization is associated with the degree of lipid order.40In this view, the presence of lipids that are, due to their shape, not ideally suited to form lipid bilayers will prevent a tight packing of the lipid headgroups and at least transiently expose regions of the hydrophobic interior, which may serve as triggers for the integration of α-synuclein oligomers and therefore lead to membrane permeabilization. In cellular membranes such characteristics are exhibited by phosphatidic acid (PA) and cardiolipin (CL). In fact, a propensity for membrane permeabilization by α-synuclein oligomers was observed in vesicular model systems containing these lipids.40 While PA is present in cellular membranes only in minute amounts, the inner membrane of mitochondria contains a high fraction of CL, which is further enriched in contact sites between the outer and inner membrane.51 As many lines of evidence suggest that an impairment of mitochondrial function (reviewed by Yao and Wood52) is associated with the development of PD, membrane permeabilization by α-synuclein oligomers at CL-rich sites may contribute to the toxic effect of these aggregates on the mitochondria.
Impact of α-synuclein monomers on the membrane permeabilization
Neurons containing α-synuclein oligomers will in all likelihood still have an excess of monomeric α-synuclein. Thus, it is of great interest to understand to what extent the monomers are able to influence the impairment of membrane integrity by α-synuclein oligomers. An attenuation of oligomer-induced membrane permeabilization has been described earlier,39 yet no kinetic data on the process are available. In our experiments we could observe that α-synuclein monomers are able to inhibit the membrane permeabilization caused by the oligomeric species. From a mechanistic point of view, the attenuation of membrane permeabilization could be caused in different ways. Besides stabilization of the membrane by the monomers, it is also conceivable that the monomers directly interact with the oligomers or sequester the negatively charged lipids, occupying binding sites of the α-synuclein oligomers. As the attenuation of the membrane permeabilization is only transient, a likely scenario is that the α-synuclein oligomers in a first slow step have to displace the monomers from the bilayer, which delays the onset of membrane permeabilization. This hypothesis is corroborated by the observation that membrane permeabilization is only attenuated for monomer concentrations corresponding to a saturation of available binding sites. This result would suggest a much higher membrane affinity of the oligomers compared to monomers, which implies that in the steady state case present in affected neurons the monomers might not be able to counteract the toxicity of α-synuclein oligomers.Kinetics of the membrane permeabilization
In order to measure the influx kinetics of the dithionite into the POPG LUVs, the C6-NBD-PC present in the outer leaflet was first bleached during a 5 min incubation period. After the subsequent addition of α-synuclein oligomers at varying concentrations, the influx of dithionite in the LUVs could be followed as a reduction of fluorescence of the NBD-labeled lipids on the inner leaflet. The loss of NBD fluorescence could be fitted with biexponential decay curves. Apart from a fast phase whose amplitude increased with the concentration of α-synuclein oligomers, but with an unchanged reaction rate constant, a second reaction with much slower, but concentration-dependent, kinetics was observed. The amplitude of this phase decreased with increasing α-synuclein oligomer concentration. α-Synuclein oligomers thus exhibit complex interactions with lipid bilayers. We postulate two different models to explain this behavior. It is conceivable that the fast loss of fluorescence is caused by a rapid influx of dithionite into a fraction of the vesicles, followed by a slower permeation of dithionite into the LUVs. Assuming a lipid headgroup size of 0.75 nm2 per POPG molecule, the inner leaflet of a LUV of 100 nm diameter would contain ∼42![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 lipid molecules. With a labeling ratio of 1 mol%, around 400 C6-NBD-PC molecules would be present. Thus, it is likely that during the initial period a sufficient number of dithionite molecules are able to enter the vesicle and completely bleach all fluorescent probes, suggesting that the increase in amplitude with the concentration of α-synuclein oligomers reflects an increase in the fraction of affected vesicles. This scenario fits very well with the observation that the rate constant is largely independent of the α-synuclein oligomer concentration, as the bleaching of C6-NBD-PC is limited only by the velocity of the bleaching reaction. Thus, a partial loss of fluorescence of single vesicles is unlikely. This conclusion is corroborated by the observation that after a 60 min preincubation with oligomers, a fast loss of fluorescence can still be observed, suggesting that the membrane permeabilization associated with the fast process is not transient. Additionally, a second process further reducing the fluorescence signal takes place. This slow process may be due to an increase in membrane permeability caused by the integration of α-synuclein oligomers into the lipid bilayer. At an α-synuclein concentration (monomer equivalent) of about 0.5 μM a plateau (0.016 min−1) of the influx rate constant is reached. Assuming an oligomer size of 25–30 monomers (unpublished data) this would correspond to an oligomer concentration of ∼10 nM. At a lipid concentration of 20 μM the LUV concentration would be about 0.25 nM, which corresponds to ∼40–80 oligomers per LUV. If all the oligomer complexes contribute equally to the influx of dithionite this would correspond to an influx with a rate constant of 0.0004 to 0.0002 min−1 per oligomer, a value that would be very slow for a channel like pore.
000 lipid molecules. With a labeling ratio of 1 mol%, around 400 C6-NBD-PC molecules would be present. Thus, it is likely that during the initial period a sufficient number of dithionite molecules are able to enter the vesicle and completely bleach all fluorescent probes, suggesting that the increase in amplitude with the concentration of α-synuclein oligomers reflects an increase in the fraction of affected vesicles. This scenario fits very well with the observation that the rate constant is largely independent of the α-synuclein oligomer concentration, as the bleaching of C6-NBD-PC is limited only by the velocity of the bleaching reaction. Thus, a partial loss of fluorescence of single vesicles is unlikely. This conclusion is corroborated by the observation that after a 60 min preincubation with oligomers, a fast loss of fluorescence can still be observed, suggesting that the membrane permeabilization associated with the fast process is not transient. Additionally, a second process further reducing the fluorescence signal takes place. This slow process may be due to an increase in membrane permeability caused by the integration of α-synuclein oligomers into the lipid bilayer. At an α-synuclein concentration (monomer equivalent) of about 0.5 μM a plateau (0.016 min−1) of the influx rate constant is reached. Assuming an oligomer size of 25–30 monomers (unpublished data) this would correspond to an oligomer concentration of ∼10 nM. At a lipid concentration of 20 μM the LUV concentration would be about 0.25 nM, which corresponds to ∼40–80 oligomers per LUV. If all the oligomer complexes contribute equally to the influx of dithionite this would correspond to an influx with a rate constant of 0.0004 to 0.0002 min−1 per oligomer, a value that would be very slow for a channel like pore.
Alternatively, the loss of fluorescence could also be explained by lipid flip-flop processes which expose yet unbleached C6-NBD-PC molecules to the dithionite. In a flip-flop assay we observed that α-synuclein oligomers enhance the lipid flip-flop in a concentration dependent fashion. For POPG LUVs preincubated with oligomers for 60 min the extent of flipped lipids reaches only about 10% for high oligomer concentrations and even decreases slightly for the highest concentrations. A leakage assay performed with LUVs preincubated with oligomers for 60 min showed that also in this case a high fraction of vesicles is permeable to dithionite, leaving only the fraction of non-permeabilized vesicles contributing to the retained fluorescence signal. As this fraction is in the range of 30–45% for the two highest oligomer concentrations measured, it can be estimated that the real extent of lipid flip-flop is much higher. Therefore, an enhanced lipid flip-flop is a likely cause for the observed slow process, where the observed loss of fluorescence is due to C6-NBD-PC becoming exposed to dithionite anions.
Material and methods
Liposome preparation
Liposome preparation was performed as described earlier.40Lipids were obtained from Avanti Polar lipids and used without further purification, other chemicals were obtained from Sigma Aldrich unless indicated otherwise. Specific lipid compositions were obtained by mixing the chloroform dissolved lipids at specific ratios. To prepare LUVs, a thin lipid film was formed by drying of 1 μmol of lipid in a glass tube using a gentle stream of N2. The lipid film was then hydrated by adding 1 ml of a solution of 150 mM KCl, 10 mM Hepes, pH 7.4. Hydration was continued with vortexing for approximately 5 minutes. The sample was subsequently subjected to 5 freeze–thaw cycles by dipping into liquid nitrogen and thawing above the lipid phase transition temperature. The resulting solution was extruded 11 times through a polycarbonate membrane filter with a 100 nm pore size. The homogeneity of the LUV population was confirmed on a regular basis by dynamic light scattering experiments.Preparation of human wildtype α-synuclein monomers and oligomers
Expression of human wild-type α-synuclein was performed as described previously,53 and the procedure for preparation of oligomers was adapted from van Rooijen et al. (2009).40 In short, to prepare the oligomeric α-synuclein species, 250 μM stock solutions of α-synuclein monomers in 10 mM Hepes pH 7.4 were dried in a vacuum evaporator and dissolved using deionized water at a protein concentration of 1 mM. The protein solution was incubated in an Eppendorf thermomixer for 18 h at room temperature (RT) and 300 rpm, followed by a 3 h incubation at 37 °C. After filtration through a 0.2 μm Spin-X centrifugal filter (Corning) to remove possible large aggregates, the oligomeric species were separated from the monomeric protein by size-exclusion chromatography on a Superdex 200 prep grade (GE) gel filtration column using 10 mM Hepes pH 7.4, 150 mM KCl as eluant. Fractions containing the αS oligomers, judged from UV absorption at 280 nm, were pooled and concentrated using a Vivaspin (Sartorius) concentrator with a 10 kDa molecular weight cutoff. The protein concentration was estimated by measuring the absorption at 280 nm. All concentrations given for oligomeric α-synuclein refer to the respective equivalent monomer concentration.Leakage assay
To measure bilayer leakage, samples containing 20 μM of LUVs in 10 mM Hepes pH 7.4, 150 mM KCl were prepared. In all cases 1 mol% of C6-NBD-PC was included. Subsequently α-synuclein oligomers and/or monomers and 1% (v/v) dithionite stock (1 M Na2S2O4 and 1 M Tris) were added at timepoints indicated in the description of the experiment, and the decrease in C6-NBD-PC fluorescence was followed in time. The loss of fluorescence is thereby caused by the reducing capacity of the dithionite anions which convert the fluorescent 7-nitro-2-1,3-benzoxadiazol-4-yl moiety of the C6-NBD-PC to the non-fluorescent 7-amino-2-1,3-benzoxadiazol-4-yl group.46 In order to follow the oligomer-facilitated influx of dithionite, 10 mM dithionite (diluted from dithionite stock) was incubated with the LUVs for 5 minutes, which resulted in a bleaching of the outer bilayer leaflet. After bleaching of the outer bilayer leaflet, oligomers (10% of total sample volume) were added to the LUVs and the C6-NBD-PC fluorescence was followed in time. In all cases before the addition of the dithionite the initial fluorescence (F0) was recorded. To determine the background fluorescence (Fbgd) the LUVs were completely disrupted after the experiment by the addition of 0.5% (v/v) Triton X-100. After background subtraction (Fbgd), the values obtained for F0 were used to normalize fluorescence intensities and account for sample to sample variations. All measurements were performed at room temperature. The NBD-fluorescence was excited with 470 nm (5 nm bandwidth) and emission was measured at 540 nm (20 nm bandwidth) on a Cary Eclipse (Varian) fluorescence spectrometer.To determine the total extent of membrane leakage the fluorescence intensity was measured 8 min after oligomer addition (F8min). The amount of unbleached C6-NBD-PC was calculated using the expression:
| [(F8min − Fbgd)/(F0 − Fbgd)] × 100 |
| Arel = A/(0.9 × F0 − Fbgd) |
Lipid flip-flop assay
C6-NBD-PC was dried as a thin film in a glass tube, and subsequently redissolved in 10 mM Hepes pH 7.4, 150 mM KCl to a final concentration of 20 μM by vortexing for several minutes. To measure lipid flip-flop, samples containing 20 μM of POPG LUVs in 10 mM Hepes pH 7.4, 150 mM KCl were prepared. C6-NBD-PC was added to the vesicles to yield a fraction of 1 mol% of total lipids. The vesicles were incubated for 5 min at RT to allow for complete insertion of C6-NBD-PC into the outer leaflet of the LUVs. Subsequently α-synuclein oligomers were added and the sample was incubated at RT for 60 min to allow for lipid flip-flop. The amount of C6-NBD-PC flipped to the inner leaflet was measured in the same way as described for the experiments measuring the total extent of membrane leakage, with the exception that the remaining fluorescence was determined already after 6 min.Conclusions
Considering the sharp cutoff in the fraction of negatively charged lipids below which membrane permeabilization is precluded and the complex kinetics of the loss of fluorescence, we suggest that the interactions between the α-synuclein oligomers and a lipid bilayer cannot be described by a simple model based on the integration of pores. A model consistent with the data on charge density and the observed influx rate constants would be that membrane bound α-synuclein oligomers strongly disturb the lipid order, creating membrane defects which allow the fast influx of dithionite. However, the strong disruption may not occur with each binding event of α-synuclein oligomers, so that only a fraction of the LUVs is affected. In addition, a second rate constant for the loss of fluorescence can be observed. This rate constant however is very slow, and may be explained by the enhancement in lipid flip-flop, exposing C6-NBD-PC from the inner leaflet of LUVs that are not permeable to dithionite. In this view, it is likely that the α-synuclein oligomers do not form distinct protein pores, but permeabilize lipid bilayers by a decrease of lipid order. This is in good agreement with the observed enhancement of lipid flip-flop and the strong correlation between membrane stability and membrane permeabilization (this work and van Rooijen et al.40).Acknowledgements
M.S. is sponsored by a German Academic Exchange Service (DAAD) postdoctoral fellowship (D/09/50722) and by NanoNextNL.References
- M. C. de Rijk, L. J. Launer, K. Berger, M. M. Breteler, J. F. Dartigues, M. Baldereschi, L. Fratiglioni, A. Lobo, J. Martinez-Lage, C. Trenkwalder and A. Hofman, Neurology, 2000, 54, S21–S23 CAS.
- J. W. Heath, Arch. Neurol Psychiatry, 1947, 58, 484–497 Search PubMed.
- K. Jellinger and A. Jirasek, Acta Neuropathol., 1971, 5(Suppl 5), 3–16.
- T. R. Scott and M. G. Netsky, Int. J. Neurol., 1961, 2, 51–60 Search PubMed.
- E. Forster and F. H. Lewy, Paralysis agitans, Springer Verlag, Berlin, 1912 Search PubMed.
- E. Ohama and F. Ikuta, Acta Neuropathol., 1976, 34, 311–319 CrossRef CAS.
- R. Kruger, W. Kuhn, T. Muller, D. Woitalla, M. Graeber, S. Kosel, H. Przuntek, J. T. Epplen, L. Schols and O. Riess, Nat. Genet., 1998, 18, 106–108 CrossRef CAS.
- M. H. Polymeropoulos, C. Lavedan, E. Leroy, S. E. Ide, A. Dehejia, A. Dutra, B. Pike, H. Root, J. Rubenstein, R. Boyer, E. S. Stenroos, S. Chandrasekharappa, A. Athanassiadou, T. Papapetropoulos, W. G. Johnson, A. M. Lazzarini, R. C. Duvoisin, G. Di Iorio, L. I. Golbe and R. L. Nussbaum, Science, 1997, 276, 2045–2047 CrossRef CAS.
- J. J. Zarranz, J. Alegre, J. C. Gomez-Esteban, E. Lezcano, R. Ros, I. Ampuero, L. Vidal, J. Hoenicka, O. Rodriguez, B. Atares, V. Llorens, E. Gomez Tortosa, T. del Ser, D. G. Munoz and J. G. de Yebenes, Ann. Neurol., 2004, 55, 164–173 CrossRef CAS.
- T. Bartels, J. G. Choi and D. J. Selkoe, Nature, 2011, 477, 107–110 CrossRef CAS.
- A. K. Frimpong, R. R. Abzalimov, V. N. Uversky and I. A. Kaltashov, Proteins, 2010, 78, 714–722 Search PubMed.
- M. M. Dedmon, K. Lindorff-Larsen, J. Christodoulou, M. Vendruscolo and C. M. Dobson, J. Am. Chem. Soc., 2005, 127, 476–477 CrossRef CAS.
- Y. H. Sung and D. Eliezer, J. Mol. Biol., 2007, 372, 689–707 CrossRef CAS.
- C. W. Bertoncini, Y. S. Jung, C. O. Fernandez, W. Hoyer, C. Griesinger, T. M. Jovin and M. Zweckstetter, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 1430–1435 CrossRef CAS.
- T. Antony, W. Hoyer, D. Cherny, G. Heim, T. M. Jovin and V. Subramaniam, J. Biol. Chem., 2003, 278, 3235–3240 CrossRef CAS.
- C. O. Fernandez, W. Hoyer, M. Zweckstetter, E. A. Jares-Erijman, V. Subramaniam, C. Griesinger and T. M. Jovin, EMBO J., 2004, 23, 2039–2046 CrossRef CAS.
- W. Hoyer, D. Cherny, V. Subramaniam and T. M. Jovin, Biochemistry, 2004, 43, 16233–16242 CrossRef CAS.
- J. Kim, R. Harada, M. Kobayashi, N. Kobayashi and K. Sode, Mol. Neurodegener., 2010, 5, 20 Search PubMed.
- I. V. Murray, B. I. Giasson, S. M. Quinn, V. Koppaka, P. H. Axelsen, H. Ischiropoulos, J. Q. Trojanowski and V. M. Lee, Biochemistry, 2003, 42, 8530–8540 CrossRef CAS.
- V. N. Uversky and A. L. Fink, Biochim. Biophys. Acta, 2004, 1698, 131–153 CAS.
- D. Eliezer, E. Kutluay, R. Bussell, Jr. and G. Browne, J. Mol. Biol., 2001, 307, 1061–1073 CrossRef CAS.
- T. S. Ulmer, A. Bax, N. B. Cole and R. L. Nussbaum, J. Biol. Chem., 2005, 280, 9595–9603 CAS.
- C. C. Jao, A. Der-Sarkissian, J. Chen and R. Langen, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 8331–8336 CrossRef CAS.
- D. F. Clayton and J. M. George, Trends Neurosci., 1998, 21, 249–254 CrossRef CAS.
- L. Maroteaux, J. T. Campanelli and R. H. Scheller, J. Neurosci., 1988, 8, 2804–2815 CAS.
- V. N. Uversky, FEBS J., 2010, 277, 2940–2953 CrossRef CAS.
- K. A. Conway, S. J. Lee, J. C. Rochet, T. T. Ding, R. E. Williamson and P. T. Lansbury, Jr., Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 571–576 CrossRef CAS.
- K. M. Danzer, D. Haasen, A. R. Karow, S. Moussaud, M. Habeck, A. Giese, H. Kretzschmar, B. Hengerer and M. Kostka, J. Neurosci., 2007, 27, 9220–9232 CrossRef CAS.
- T. F. Outeiro, P. Putcha, J. E. Tetzlaff, R. Spoelgen, M. Koker, F. Carvalho, B. T. Hyman and P. J. McLean, PLoS One, 2008, 3, e1867 CrossRef.
- B. Winner, R. Jappelli, S. K. Maji, P. A. Desplats, L. Boyer, S. Aigner, C. Hetzer, T. Loher, M. Vilar, S. Campioni, C. Tzitzilonis, A. Soragni, S. Jessberger, H. Mira, A. Consiglio, E. Pham, E. Masliah, F. H. Gage and R. Riek, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 4194–4199 CrossRef CAS.
- B. Caughey and P. T. Lansbury, Annu. Rev. Neurosci., 2003, 26, 267–298 CrossRef CAS.
- M. J. Volles and P. T. Lansbury, Jr., Biochemistry, 2003, 42, 7871–7878 CrossRef CAS.
- M. R. Cookson and O. Bandmann, Hum. Mol. Genet., 2010, 19, R21–R27 CrossRef CAS.
- P. K. Auluck, G. Caraveo and S. Lindquist, Annu. Rev. Cell Dev. Biol., 2010, 26, 211–233 CrossRef CAS.
- A. M. Cuervo, E. S. Wong and M. Martinez-Vicente, Mov. Disord., 2010, 25(Suppl 1), S49–S54 CrossRef.
- D. Sulzer, Mov. Disord., 2010, 25(Suppl 1), S27–S31 CrossRef.
- L. R. Feng, H. J. Federoff, S. Vicini and K. A. Maguire-Zeiss, Eur. J. Neurosci., 2010, 32, 10–17 CrossRef.
- M. Zhu, J. Li and A. L. Fink, J. Biol. Chem., 2003, 278, 40186–40197 CrossRef CAS.
- M. J. Volles and P. T. Lansbury, Jr., Biochemistry, 2002, 41, 4595–4602 CrossRef CAS.
- B. D. van Rooijen, M. M. Claessens and V. Subramaniam, Biochim. Biophys. Acta, 2009, 1788, 1271–1278 CAS.
- M. Stöckl, P. Fischer, E. Wanker and A. Herrmann, J. Mol. Biol., 2008, 375, 1394–1404 CrossRef.
- E. Rhoades, T. F. Ramlall, W. W. Webb and D. Eliezer, Biophys. J., 2006, 90, 4692–4700 CrossRef CAS.
- B. D. van Rooijen, M. M. Claessens and V. Subramaniam, FEBS Lett., 2008, 582, 3788–3792 CrossRef CAS.
- H. A. Lashuel, B. M. Petre, J. Wall, M. Simon, R. J. Nowak, T. Walz and P. T. Lansbury, Jr., J. Mol. Biol., 2002, 322, 1089–1102 CrossRef CAS.
- T. T. Ding, S. J. Lee, J. C. Rochet and P. T. Lansbury, Jr., Biochemistry, 2002, 41, 10209–10217 CrossRef CAS.
- J. C. McIntyre and R. G. Sleight, Biochemistry, 1991, 30, 11819–11827 CrossRef CAS.
- W. S. Davidson, A. Jonas, D. F. Clayton and J. M. George, J. Biol. Chem., 1998, 273, 9443–9449 CrossRef CAS.
- E. Jo, J. McLaurin, C. M. Yip, P. St. George-Hyslop and P. E. Fraser, J. Biol. Chem., 2000, 275, 34328–34334 CrossRef CAS.
- M. Ramakrishnan, P. H. Jensen and D. Marsh, Biochemistry, 2003, 42, 12919–12926 CrossRef CAS.
- B. D. van Rooijen, K. A. van Leijenhorst-Groener, M. M. Claessens and V. Subramaniam, J. Mol. Biol., 2009, 394, 826–833 CrossRef CAS.
- D. Ardail, J. P. Privat, M. Egret-Charlier, C. Levrat, F. Lerme and P. Louisot, J. Biol. Chem., 1990, 265, 18797–18802 CAS.
- Z. Yao and N. W. Wood, Antioxid. Redox Signaling, 2009, 11, 2135–2149 Search PubMed.
- M. E. van Raaij, I. M. Segers-Nolten and V. Subramaniam, Biophys. J., 2006, 91, L96–L98 CrossRef CAS.
Footnotes |
| † Published as part of a Molecular BioSystems themed issue on Intrinsically Disordered Proteins: Guest Editor M. Madan Babu. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c1mb05293d |
| This journal is © The Royal Society of Chemistry 2012 |