Intrinsic disorder in the androgen receptor: identification, characterisation and drugability†
Iain J.
McEwan
School of Medical Sciences, University of Aberdeen, IMS Building, Foresterhill, Aberdeen AB25 2ZD, Scotland, UK. E-mail: iain.mcewan@abden.ac.uk; Tel: + 44 (0)1224 437328
First published on 5th August 2011
Abstract
The androgen receptor (AR) regulates networks of genes in response to the steroid hormones testosterone and dihydrotestosterone. The receptor protein is made up of both stably folded globular domains, involved in hormone and DNA binding, and regions of intrinsic disorder, including the N-terminal domain (NTD). The AR-NTD has a modular activation function (termed AF1) and is important for gene regulation, participating in multiple protein-protein interactions. Biophysical studies have revealed that AR-NTD/AF1 has limited stable secondary structure and conforms to a ‘collapsed disordered’ conformation. The AR-NTD/AF1 has the propensity to adopt an α-helical conformation in response to a natural osmolyte or a co-regulatory binding partner. The AR is a key drug target in the management of advanced prostate cancer and recently a small molecule inhibitor was identified that interacts with the NTD/AF1 and impairs protein-protein interactions and recruitment of the receptor to target genes. In this review the role of intrinsic disorder in AR function is discussed along with the potential to develop new drugs that will target the structurally plastic NTD.
 Iain J. McEwan |
1. Introduction
“Confusion heard his voice, and wild uproar Stood ruled, stood vast infinitude confined; Till at his second bidding darkness fled, Light shone, and order from disorder sprung.”John Milton, Poet
“However, it required some years before the scientific community in general accepted that flexibility and disorder are very relevant molecular properties also in other systems.”
Robert Huber, Biochemist and Nobel Laureate
The steroid hormones testosterone and dihyrdotestosterone are small, four ringed planar molecules that exert profound affects important for the development of the male phenotype during embryogenesis and at puberty.1Androgen signalling is also important for reproductive health in both male and females as well as anabolic actions in bone and muscle tissue (see ref. 2, 3 and references therein). The actions of these potent signalling molecules are mediated by the product of a single gene that codes for a protein of 110 kDa: the androgen receptor (AR; NR3C4) is an intracellular protein that functions as a ligand-activated transcription factor (Fig. 1).4 The AR is a member of the nuclear receptor superfamily, which in addition to steroid receptors includes receptors for vitamins, bile acids and fatty acid derivatives.5,6 These proteins are highly modular and contain a ligand binding domain (LBD) in the C-terminus, a flexible hinge region linking to the DNA binding domain (DBD) and then a highly variable N-terminal domain (NTD) (Fig. 2A).6
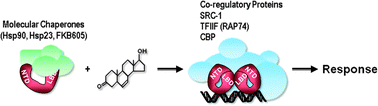 | ||
| Fig. 1 Androgen Receptor mechanism of Action. In the absence of the hormone the AR is in a complex with molecular chaperones and immunophilin proteins.79 Binding of testosterone causes a conformational change in the receptor protein, nuclear translocation and binding to target genes. Interactions with the transcriptional machinery and histone modifying enzymes (TFIIF, CBP, SRC1) results in activation of networks of target genes and the cellular response to hormone. | ||
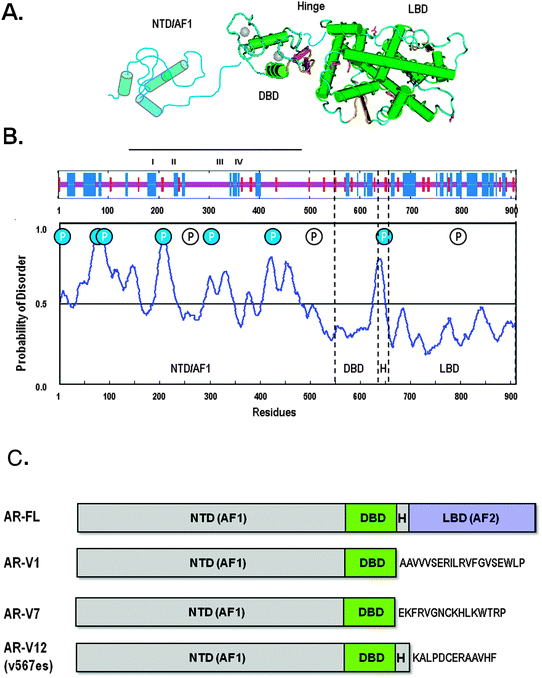 | ||
| Fig. 2 Domain Organisation and Structure of the Androgen Receptor and Splice Variants. (A) Schematic representation of the AR protein showing the 3-D structure for the isolated ligand (LBD) and DND (DBD) binding domains, together with representation of the structurally flexible hinge and N-terminal domain (NTD). (B) Prediction of secondary structure content in the human AR (upper panel) and regions of intrinsic disorder (RONN plot; lower panel). The Blue and red bars represent α-helix and β-strand structure respectively and the location of four putative helices in AR-AF1 (line, I–IV) are indicated. In the RONN plot peaks represent high probability (> 0.5) of intrinsic disorder. Also shown on this plot is the occurrence of known phosphorylated serine residues: filled circles indicate those residues that are found in regions of intrinsic disorder. (C) Schematic drawing of the full-length AR and representative N-terminal splice variants. | ||
2. Androgen receptor organisation
2.1 Ligand binding domain (LBD)
The LBD is built-up of approximately 12 α-helices arranged in three layers forming a sandwiched like structure, with the ligand binding pocket (LBP) buried in the interior.7–9 The binding of steroid leads to a rearrangement of helix 12 at the C-terminal end of the protein forming a lid over the LBP. The conformational change also creates a surface on the LBD, termed activation function2 (AF2) that is important for protein-protein interactions with co-regulatory proteins. The AF2 is made up of residues from helices 3, 4, 5 and 12 which form a hydrophobic pocket that interacts with FxxLF or the related LxxLL peptide motifs.7,10 The AF2 surface on the AR-LBD is unique in preferring to interact with the more bulky hydrophobic residues and significantly a FxxLF sequence in the receptor NTD. This N/C-terminal interaction has been shown to be functionally important for selective AR-dependent gene expression. However, interestingly, this N/C terminal interaction may be disrupted by DNA binding, which in turn would expose surfaces within the NTD and the AF2 pocket for interactions with co-regulatory proteins.11 A second surface on the AR-LBD, termed BF3, has also been described which can bind small molecules (see below) and allosterically regulate protein-protein interactions at AF2.9In other nuclear receptors, for example oestrogen (ER; NR3A1) and retinoid X (RXR; NR2B1) receptors, sequences in helix 10/11 have been shown to represent a dimerization surface. However, whither this is true for the AR is a subject of debate and it is noteworthy that in the crystal structures of the AR-LBD only monomers were observed. A recent comparison of the AR-LBD with the glucocorticoid receptor (GR; NR3C1) sequence and structure identified residues in helices 1, 3 and 5 as a potential dimerization interface.12 Additional experiments attempting to disrupt potential dimerization interfaces will be required to resolve this question.
2.2 DNA binding domain (DBD)
The DBD consists of approximately 66 amino acids arranged as two Zn-finger-like structures, where the Zn ions are co-ordinated by four cysteine residues. The domain folds into a compact globular structure characterised by two perpendicular α-helices, one of which forms the recognition helix which sits in the major groove of the DNA.13 Three residues that make up the P-box, Gly577, Ser578 and Val581, have been shown to form both direct and water mediate hydrogen bonds with the nucleotides in the DNA binding site.13Steroid receptors, such as the AR, are thought to bind to DNA as homo-dimers and a 15 bp palindromic hormone response element (HRE) has been described: 5′-GGA/TACAnnnTGTTCT-3′. This canonical HRE is also recognised by the GR, progesterone (PR; NR3C3) and mineralocorticoid (MR; NR3C2) receptors.14DNA sequences selective for the AR, termed androgen response elements (ARE), have been described, which can be read as a partial palindromic or direct repeat of the 6 bp half site.15 Recent genome-wide ChIP-chip or ChIP-seq analysis of AR regulated genes in cultured cells or tissues has revealed that receptor binding sites show considerable variation in response element architecture, including imperfect palindromic sequences and half-sites in addition to clearly recognizable AREs.16–19 From several of these studies the following consensus response element can be determined: 5′-A/GGAACAnnnTGTT/GCC/T-3′ (The size of the letter is an approximate representation of the occurrence of each nucleotide at that position; McEwan unpublished). Another feature to emerge from these studies is that hormone binding sites occur within 50 kb of the start site of the target gene and are composite sites with binding sites for other transcription factors, for example FoxA1, GATA2, Oct1, AP1, HNF4α and ETS1.16–19 However, a number of outstanding questions remain: what roles do other transcription factors and co-regulatory proteins play in the tissue specific expression of AR-regulated genes; and what are the mechanisms that underlie changes in the pattern of AR regulated genes in diseases, such as prostate cancer?In the second Zn-finger a sequence of five amino acids (Ala596-Ser-Arg-Asn-Asp600) makes up the D-box and represent a dimerization interface between the two receptor monomers.13 Evidence from work with other steroid receptors suggests that dimerization occurs on the DNA and initial binding involves a receptor monomer. Work with the glucocorticoid receptor also suggests that DNA can act as an allosteric regulator of receptor function, such that binding to different DNA sequences causes changes in surfaces of the DBD that serve as binding sites for co-regulator proteins.20 Furthermore, binding of the co-regulatory protein JPD2 to the PR-DBD leads to increased helical secondary structure in the receptor NTD.21 Collectively the evidence suggest that the DBD can have a more active role in regulating receptor function than simply tethering the receptor dimer to DNA.
2.3 N-terminal domain (NTD)
In contrast to the LBD and DBD there is little or no sequence homology between the AR-NTD and the NTD of other steroid, or indeed nuclear, receptor.4,22,25 The length of the NTD is also highly variable between different family members, with the AR, PR and MR having the largest NTDs. The AR-NTD is characterized by the presence of several amino acid repeats including two large polymorphic glutamine and glycine stretches of 20 to 23 residues and 16 to 23 residues respectively; two shorter glutamine repeats of 5 and 6 residues; and short repeats of 5 alanine and 8 proline residues; and a repeat of the sequence PSTLTL.22–25 Expansion of the large poly-glutamine repeat is associated with Kennedy's disease or spinal bulbar muscular atrophy a neuromuscular degenerative disease affecting the motor neurones in the brain stem and spinal column (ref. 23 and references therein). There are three regions within the AR-NTD that show high homology between the receptor from different species, these are amino acids 1 to 25, 224 to 258 and 500 to 541, directly adjacent to the DBD.25Earlier work has shown that deletion of the NTD resulted in a transcriptional weak or silent receptor protein.26,27 And sequences within the NTD have been delineated that are important for receptor-dependent gene regulation and these have been termed collectively activation function 1 (AF1). Brinkmann and co-workers highlighted two regions they termed TAU1 (amino acids 101 to 370) and TAU5 (amino acids 360 to 485), which overlapped with the region identified by Wilson and co-workers (amino acids 141 to 338).26,27 Significantly, Heet al.7 demonstrated using Gal4 fusion constructs, that the length of the NTD correlated with the importance of AF1 for nuclear receptor function.
The first co-regulatory protein identified binding directly to the AF1 region of the AR was the large subunit, RAP74, of the general transcription factor TFIIF.28TFIIF is important in the recruitment of RNA polymerase II to the start site of transcription and in regulating the elongation activity of the polymerase. Further work identified receptor binding sites within the globular N- and C-terminal domains of RAP74, which bound AR-AF1 with relative binding affinities of 0.29 μM and 0.69 μM respectively.24,29 Subsequently an ever growing list of binding partners has been described, including the co-activators SRC-1, CBP and ART27 (reviewed in ref. 30). Collectively, these studies emphasise the importance of regions in the AR-NTD for communicating with the cell's transcriptional machinery. An intriguing question now is whether different surfaces in the AF1 or the AF2 pocket in the LBD mediate differential gene expression. Yamamoto and co-workers31 identified a number of glucocorticoid-regulated genes that depended selectively on either GR- AF1 or -AF2 surfaces for induction, but this remains to be comprehensively studied for the AR and other steroid receptors.
3. Intrinsic disorder in the AR
3.1 Hinge region
The hinge region is a flexible link between the DBD and the LBD and spans amino acids 624 to 693. The hinge region contains the C-terminal extension (CTE) of the DBD, NLS and sites for post-translational modifications. The CTE has been shown to be important for AR selective response element recognition and binding and appears disordered in structural analysis of the isolated DBD of the AR, glucocorticoid receptor and oestrogen receptors (ref. 13 and references therein). Structural and functional analysis of the NLS showed that the AR NLS interacted with α-importin similar to a monopartite NLS with residues 629 to 634 binding to the armadillo repeats 2, 3 and 4.32 However, the structure the extended peptide (amino acids 617 to 634) adopted was β-turn-like and unique to the AR NLS.32Interestingly in recent structural studies of full-length nuclear receptors or two-domain receptor constructs different arrangements of the hinge region were observed. In the crystal structure of a complex of PPARγ and RXR bound to a DR1 DNA response element the PPAR monomer adopted a ‘closed’ conformation with extensive interactions between the LBD and the PPAR-DBD and the RXR-LBD, Hinge and DBD.33 In contrast the RXR monomer had an ‘open’ conformation with the hinge region extended and creating a surface for the PPAR binding. The hinge region of the PPAR also made contact with the DNA and contained 2 α-helices, while the RXR hinge region was more flexible and lacked secondary structure.33 However, these different conformations were not observed in recent solution structure studies of a number of class II receptors, which form heterodimers with RXR, bound to different response elements; although inter domain interactions between the DBD and LBD were observed.34,35 While these studies lack atomic resolution they generally all show the hinge regions in extended conformations permitting the ordering of the LBDs over the 5′ half-site of the DNA element.34
To-date a structure for a full-length, or two domain, steroid receptor complex has not been reported and it will be interesting to see how homodimerisation on palindromic-like response elements affects the architecture of the LBD and hinge region for this class of receptor.
3.2 Structural properties of the NTD
While both the isolated LBD and DBD of the AR exhibit compact α-helical globular structures the NTD lacks stable secondary structure and conforms to the concept of an intrinsically disordered protein domain (Fig. 2B). The AR- AF1 domain has 13% helical secondary structure as determined by circular dichroism, FTIR-spectroscopy and secondary structure prediction analysis.36,37 Significantly, the AR-AF1 domain has the propensity to form helical structures in the presence of a natural osmolyte, trimethylamine N-oxide (TMAO), or a hydrophobic organic solvent, trifluoroethanol (TFE), or the binding partner TFIIF (RAP74)36,37 (Fig. 3). In the presence of TMAO or RAP74-CTD there was a 3-fold increase in α-helix secondary structure.37TMAO also reduces limited proteolysis of the polypeptide and results in the two tryptophan residues within AF1 becoming less solvent exposed, which is consistent with folding or increased conformational stability of the receptor domain.38 Four predicted α-helices within AF1 (Fig. 2B, upper panel) were disrupted by introducing helix breaking proline residues.36 More recently, we could show that AR-AF1 had structural properties consistent with a molten-globule like conformation that has been referred to as ‘collapsed disorder’.38 Current models for the AR-AF1 domain suggest that it exists as an ensemble of conformations with limited secondary and tertiary structure, which are primed to bind co-regulatory proteins. Binding of co-regluatory partner proteins, and possibly DNA response elements, stabilises a particular conformation from this ensemble or involves induced folding of the ensemble to a more helical conformation (Fig. 3).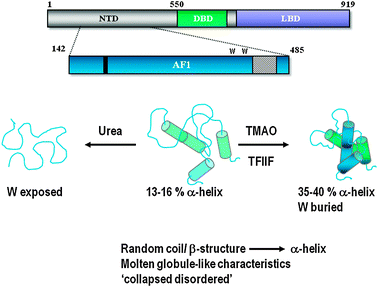 | ||
| Fig. 3 Regulation of AR-NTD Folding. The AR-NTD/AF1 is proposed to exist as an ensemble of conformations having limited stable structure, described as a ‘collapsed disordered’ conformation. However, this domain will adopt a more helical conformation in the presence of the natural osmolyte TMAO or a co-regulatory binding protein (i.e.TFIIF). Folding or stabilization of the AR-AF1/NTD conformation may also be regulated by DNA binding and post-translational modifications (see text for full discussion). | ||
Interestingly the NTD has been found to modulate the DNA binding affinity of the AR-DBD, which was dependent upon the two domains being covalently bound, suggesting intradomain communication.39 The NTD reduced the binding affinity for a general HRE to a greater degree than AREs and so may act to minimise non-specific interactions of the receptor with DNA.39 Alternatively, the affect of the NTD on DNA-binding was a consequence of the absence of the LBD and resulted in steric effects that would be countered by the AR-LBD. Further experiments will be needed to resolve these possibilities.
Analysis of the full-length AR-NTD reveals similar structural flexibility and propensity to form helical conformations, suggesting the results for AF1 are not a consequence of using a fragment of the NTD. Deleting the large poly-glutamine repeat within the AR-NTD resulted in a polypeptide with less helical content and more β-structure, which showed a marked reduction in forming α-helical structure in a hydrophobic environment.23 In contrast the expanded poly-Q repeat from a patient with Kennedy's disease (Q45) showed a modest increase in α-helix and non-ordered structure, but retained the propensity to adopt significant helical conformation at the expense of β-structure and non-ordered structure in the presence of TFE.23 Collectively these findings suggest that the poly-Q repeat is important for modulating the structural plasticity of the AR-NTD/AF1 domain.
It was striking in the recent crystal structure of the PPARγ-RXR complex that the electron density for the receptor NTDs was missing and in hydrogen exchange experiments the highly dynamic nature of these regions was demonstrated.33 It seems certain therefore that the structural plasticity observed for the AR-AF1/NTD is not simply a consequence of removing the domain from the context of the full-length receptor. However, it is reasonable to expect that other regions of the AR may modulate the folding and/or stability of the AR-AF1/NTD, given the documented intradomain communications of the AR-NTD/LBD and NTD/DBD.
3.3 Post-translational modifications and intrinsically disordered regions of the androgen receptor
The prevalence of intrinsically disordered regions and protein domains in eukayotic signalling proteins and transcription factors has been attributed to the advantages of coupling induced folding with functional interactions40–42 and allosteric regulation.43 It has previously been proposed that one advantage of intrinsic disorder is permitting access for post-translational modifying enzymes.40,42 The AR is the target for a plethora of post-translational modifications, including phosphorylation, acetylation, and sumoylation (see ref. 5 and references therein). A significant number of the residues targeted for different modifications occur in regions of intrinsic disorder: namely the hinge and NTD.The AR is a phosphoprotein with up to ten phosphorylated serine residues being identified in cell culture systems44,45or more recently in vivo2,46,47 Strikingly, 70% of these serine residues are found in regions strongly predicted to be intrinsically disordered (Fig. 2B). One of these residues, serine 650, is found in the hinge domain and has been implicated in cytoplasmic/nuclear shuttling of the AR. In this same region of the receptor lysine residues that make up the NLS have been found to be acetylated48–51 and more recently methylated.52,53 These modifications have a complex effect on AR signalling. Acetylation of the lysines in the K630LKK633 sequences is important for receptor-dependent gene regulation48–50 and mutating these residues impirs activation, enhances recruitment of co-repressors,49 impairs phosphorylation of serine 94 in the NTD54and impairs nuclear translocation.55Methylation of lysines in this sequence also enhanced receptor-dependent transactivation by increasing N/C-terminal interactions and recruitment of the receptor to target genes.52,53
In the case of the ERβ56 and the GR57,58 phosphophorylation was associated with conformational changes in the regions of the AF1. Preliminary analysis of the AR-AF1 suggests that phosphorylation may also modulate folding of this domain such that the polypeptide becomes more sensitive to proteolytic challenge when phosphorylated (McEwan and co-workers unpublished data). In this regard it is interesting that inhibition of signalling cascades, impaired kinase activity or mutating phosphorylated residues results in stabilization of the receptor protein (ref. 59; McGuinness and McEwan unpublished observations). The challenge now is to identify specific modifications with domain folding/unfolding and to also indentify the potential signalling cross-talk between different post-translational modifications.
In addition to phosphorylation there are two lysine residues in the AR-NTD, at positions 385 in AF1 and 511, that are modified by sumolyation.60,61 These lysine residues fall within a sequence that has been termed a ‘synergy control’ motif, which is found in other steroid receptors (e g.GR) and transcriptional factors (e g. ETS-1).62 Mutating the lysines, in particular lysine 385, resulted in increased AR-dependent transcription at multiple HREs,61,62 but not AREs.61 The conclusion from such studies is that sumoylation of the AR-NTD mediates control of receptor cooperativity and that the architecture of the DNA binding sites can fine tune this control. The impact if any, of sumoylation on AR-NTD/AF1 folding and conformation remains to be investigated.
4. The androgen receptor as a drug target
Given the prominence of steroid hormones in normal development and physiology the receptor proteins have long been recognised as important and validated drug targets. Hirsutism and acne are conditions resulting from androgen excess and common indicators of polycystic ovarian syndrome (PCOS) while the normal androgen regulation of the prostate gland is hijacked in the growth and progression of prostate cancer. Treatment of these conditions can involve inhibiting production of androgenic steroids by the testis, or ovaries in the case of PCOS, and in the case of advanced prostate cancer using antiandrogens to block AR function.634.1 LBD
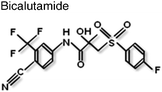
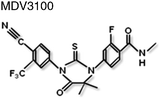
Non-steroidal antiandrogens such as flutamide and bicalutamide bind to the ligand binding pocket in the LBD and prevent agonist binding and impair AR action (Table 1). However, these antagonists can exhibit partial agonist activity as a result of over expression of the AR protein64 or point mutations in the LBD that broaden hormone specificity (see65). More recently, ‘second-generation’ AR antagonists, such as MDV3100 (Table 1), have been development, which have proved more effective at blocking AR translocation into the nucleus and thereby inhibiting receptor-dependent gene expression.66,67| Compound | Site of binding | Mechanism of inhibition | Ref. |
|---|---|---|---|
| EGCG = epigallocatechin-3-gallate (polyphenol found in Green Tea); LBD, DBD and NTD = ligand binding, DNA binding and N-terminal domains of the AR respectively; LBP = ligand binding pocket.; TRIAC = 3,3′,5-triodothyroacetic acid; ? = binding site or mechanism remains to be determined. | |||
|
LBD/LBP | Conformational change in hinge region; recruitment of co-repressors. | 80 |
|
LBD/B-Site | Disruption of AF2 surface; recruitment of co-repressors? | 67,81 |
|
LBD/LBP | Blocks receptor translocation to the nucleus. | 66 |
|
LBD | Blocks receptor translocation to the nucleus. | 82 |
|
LBD/BF3 | Allosteric disruption of AF2 surface. | 9 |
|
LBD/BF3 | Allosteric disruption of AF2 surface. | 9 |
| EGCG | LBD/LBP | Disrupts N/C-interaction, increases receptor turnover. | 69 |
|
NTD | ? | 72 |
|
NTD/AF1 | Reduces recruitment of the AR to target genes and disrupts protein-protein interactions. | 73 |
| Polyamide | — | Binds to androgen response element half-site and inhibition of DNA binding. | 83 |
|
? | Inhibition of protein-protein interactions. | 84 |
|
? | Inhibition of DNA binding. | 84 |
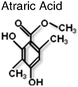
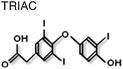
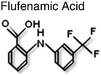
Intriguingly there are an increasing number of compounds, including natural products, which have been described as inhibiting receptor function, such as atraic acid,68polyphenols from Green Tea69 and ligands for thyroid hormone receptor, TRIAC9 (Table 1). Interestingly, the later along with flufenamic acid (Table 1) was found not to bind in the LBP, but to a discrete surface of the LBD, that was termed BF3. These small molecules were found to inhibit protein-protein interactions at the AF2 surface suggesting allosteric regulation between the two surfaces on the LBD.9
4.2 DBD
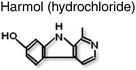
Although no small molecule inhibitors for the DBD have been described to date Dervan and co-workers35 demonstrated that polyamide molecules designed to the DNA response element half-site were able to inhibit AR activity and change the pattern of gene expression in prostate cancer cells in culture (Table 1). The polyamide is thought to work by binding to the DNA target sequence and inhibiting protein-DNA interactions.Diamond and co-workers70 have identified several compounds from libraries of compounds and natural products which disrupted the AR-NTD/LBD (CTD) interaction in a ligand screen. The two compounds (Table 1) with highest potency appeared to act at different steps in the receptor signalling pathway: one of the compounds, harmol hydrochloride, acted to inhibit binding of the AR to DNA response elements.70 However, the binding site for this molecule has not been identified.
4.3 NTD
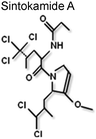
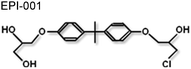
Until recently the structural plasticity of the NTD may have appeared to be a rather unattractive target for small molecule inhibitors of AR function. However, it is important to remember that this region of the receptor is crucial for function (see above) and using the intact NTD as a ‘decoy’ molecule Sadar and co-workers could demonstrate it was possible to modulate AR activity by sequestering co-regulatory proteins.71 This group has also shown that compounds isolated from a marine sponge, sintokamides A to E, which are chlorinated peptides (Table 1), targeted the NTD and inhibited receptor-dependent gene expression.72 It is therefore of particular significance that a bisphenol A ester derivative (BADGE), isolated from another species of sponge, was identified as a small molecule inhibitor of AR signalling.73 This molecule, called EPI-001 (Table 1), selectively inhibited the AR-dependent gene regulation in cell culture studies and an impressive ability to shrink tumour volume in two animal models of prostate cancer.73EPI-001 interacted with the NTD/AF1 and altered the steady state fluorescence emission spectrum for AF1 in the tyrosine region and selectively inhibited protein-protein interaction, between the AR and TFIIF (RAP74 subunit) and the coactivator CBP, in vitro.73 Overall this study provides ‘proof-of-principle’ that small molecule inhibitors can be identified which target the AR-NTD.
Recently a number of splice variants of the AR have been described lacking the LBD, LBD-Hinge or LBD-Hinge-DBD, and the most prevalent ones found in prostate cancer cells have the NTD and DBD (Fig. 2C) and these have the potential to act as constitutive transcriptional activators.74–78 In particular V7 and V12 appear to be over represented in metastatic clinical samples.75,77 While most splice variants have been observed as putative transcripts, by RT-PCR, the V1 and V7 have been identified at the protein level.74–77 It remains to be determined the exact role of these splice variants: do they act as constitutive activators of a subset of androgen regulated genes and/or as modulators of the activity of the full-length receptor? For example V7 and V12 have been shown to be nuclear and have constitutive activity,74,76 in the receptor negative cell lines PC3 and/or DU145, while V1 was found to be cytoplasmic and had conditional activity76 in the LNCaP cell line.
The advantages for targeting the NTD therapeutically is that such drugs would complement existing antiandrogens as combined therapies and critically should be effective in castrate resistant prostate cancer (CRPC) where the efficacy of antiandrogens has been compromised through amplification of the AR gene or point mutations in the protein or as result of AR splice variants, which lack the LBD.
5. Conclusions and future perspectives
The cDNA for the human AR was cloned over 20 years ago and the intervening period has seen considerable progress in our understanding of the structure-function relationships of this important ligand-regulated transcription factor and its action in different cell types of the reproductive system. This has included the functional analysis of the isolated receptor domains (NTD, DBD and LBD), the solving at atomic resolution of the structure of the DBD and LBD, the identification of binding partners and a clearer appreciation of the role of intrinsic disorder in receptor-dependent gene regulation. Also recent years has seen a significant increase in the identification of androgen regulated genes and the genomic elements that bind the receptor and the cellular functions of the receptor through targeted knock-out studies. However, despite the progress made, a number of questions remain unanswered:What is the structure of the full-length or a two-domain AR polypeptide, bound to different DNA response elements?
What conformation(s) does the AR-NTD adopt in response to different binding partners or intracellular conditions?
How are allosteric signals transmitted within or between the different receptor domains; and what role does intrinsic disorder play in this regulation?
How are different post-translational modifications integrated in a cell-specific manner?
How is the AR gene regulated in a temporally and spatially during development and in adult tissues?
In conclusion, it is assured there will be further dramatic developments as researchers strive to understand the biology and physiological actions of the androgen receptor, the structural and biochemical basis for receptor function and the impact genetic alterations have on receptor-dependent diseases such as prostate cancer.
Acknowledgements
Research in the author's laboratory is currently supported by the Chief Scientists Office, Scottish Government; The Kosterlizt centre, University of Aberdeen; and the Prostate Cancer Charity.References
- M. T. Patrao, E. J. Silva and M. C. Avellar, Arq. Bras. Endocrinol. Metabol., 2009, 53, 934–945 CrossRef.
- I. J. McEwan, D. McGuinness, C. W. Hay, R. P. Millar, P. T. Saunders and H. M. Fraser, Reproduction, 2010, 140, 93–104 CrossRef CAS.
- S. C. Manolagas, S. Kousteni and R. L. Jilka, Recent Prog. Horm. Res., 2002, 57, 385–409 CrossRef CAS.
- J. McEwan, Endocr. Relat. Cancer, 2004, 11, 281–293 CrossRef.
- I. J. McEwan, Essays Biochem., 2004, 40, 1–10 CAS.
- I. J. McEwan, Methods Mol. Biol., 2009, 505, 3–18 CAS.
- B. He, R. T. Gampe Jr, A. J. Kole, A. T. Hnat, T. B. Stanley, G. An, E. L. Stewart, R. I. Kalman, J. T. Minges and E. M. Wilson, Mol. Cell, 2004, 16, 425–438 CrossRef CAS.
- P. M. Matias, P. Donner, R. Coelho, M. Thomaz, C. Peixoto, S. Macedo, N. Otto, S. Joschko, P. Scholz, A. Wegg, S. Basler, M. Schafer, U. Egner and M. A. Carrondo, J. Biol. Chem., 2000, 275, 26164–26171 CrossRef CAS.
- E. Estebanez-Perpina, L. A. Arnold, P. Nguyen, E. D. Rodrigues, E. Mar, R. Bateman, P. Pallai, K. M. Shokat, J. D. Baxter, R. K. Guy, P. Webb and R. J. Fletterick, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 16074–16079 CrossRef CAS.
- H. J. Dubbink, R. Hersmus, C. S. Verma, H. A. van der Korput, C. A. Berrevoets, J. van Tol, A. C. Ziel-van der Made, A. O. Brinkmann, A. C. Pike and J. Trapman, Mol. Endocrinol., 2004, 18, 2132–2150 CrossRef CAS.
- M. E. van Royen, S. M. Cunha, M. C. Brink, K. A. Mattern, A. L. Nigg, H. J. Dubbink, P. J. Verschure, J. Trapman and A. B. Houtsmuller, J. Cell Biol., 2007, 177, 63–72 CrossRef CAS.
- M. M. Centenera, J. M. Harris, W. D. Tilley and L. M. Butler, Mol. Endocrinol., 2008, 22, 2373–2382 CrossRef CAS.
- P. L. Shaffer, A. Jivan, D. E. Dollins, F. Claessens and D. T. Gewirth, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 4758–4763 CrossRef CAS.
- F. Claessens and D. T. Gewirth, Essays Biochem., 2004, 40, 59–72 CAS.
- E. Schoenmakers, G. Verrijdt, B. Peeters, G. Verhoeven, W. Rombauts and F. Claessens, J. Biol. Chem., 2000, 275, 12290–12297 CrossRef CAS.
- E. C. Bolton, A. Y. So, C. Chaivorapol, C. M. Haqq, H. Li and K. R. Yamamoto, Genes Dev., 2007, 21, 2005–2017 CrossRef CAS.
- S. M. Eacker, J. E. Shima, C. M. Connolly, M. Sharma, R. W. Holdcraft, M. D. Griswold and R. E. Braun, Mol. Endocrinol., 2007, 21, 895–907 CrossRef CAS.
- C. E. Massie, B. Adryan, N. L. Barbosa-Morais, A. G. Lynch, M. G. Tran, D. E. Neal and I. G. Mills, EMBO Rep., 2007, 8, 871–878 CrossRef CAS.
- Q. Wang, W. Li, X. S. Liu, J. S. Carroll, O. A. Janne, E. K. Keeton, A. M. Chinnaiyan, K. J. Pienta and M. Brown, Mol. Cell, 2007, 27, 380–392 CrossRef CAS.
- S. H. Meijsing, M. A. Pufall, A. Y. So, D. L. Bates, L. Chen and K. R. Yamamoto, Science, 2009, 324, 407–410 CrossRef CAS.
- S. E. Wardell, S. C. Kwok, L. Sherman, R. S. Hodges and D. P. Edwards, Mol. Cell. Biol., 2005, 25, 8792–8808 CrossRef CAS.
- C. S. Choong, J. A. Kemppainen and E. M. Wilson, J. Mol. Evol., 1998, 47, 334–342 CrossRef CAS.
- P. Davies, K. Watt, S. M. Kelly, C. Clark, N. C. Price and I. J. McEwan, J. Mol. Endocrinol., 2008, 41, 301–314 CrossRef CAS.
- J. Reid, I. Murray, K. Watt, R. Betney and I. J. McEwan, J. Biol. Chem., 2002, 277, 41247–41253 CrossRef CAS.
- R. Betney and I. J. McEwan, J. Mol. Endocrinol., 2003, 31, 427–439 CrossRef CAS.
- J. A. Simental, M. Sar, M. V. Lane, F. S. French and E. M. Wilson, J. Biol. Chem., 1991, 266, 510–518 CAS.
- G. Jenster, H. A. van der Korput, J. Trapman and A. O. Brinkmann, J. Biol. Chem., 1995, 270, 7341–7346 CrossRef CAS.
- I. J. McEwan and J. Gustafsson, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 8485–8490 CrossRef CAS.
- D. N. Lavery and I. J. McEwan, Biochemistry, 2008, 47, 3352–3359 CrossRef CAS.
- D. N. Lavery and I. J. McEwan, Biochem. J., 2005, 391, 449–464 CrossRef CAS.
- I. Rogatsky, J. C. Wang, M. K. Derynck, D. F. Nonaka, D. B. Khodabakhsh, C. M. Haqq, B. D. Darimont, M. J. Garabedian and K. R. Yamamoto, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 13845–13850 CrossRef CAS.
- M. L. Cutress, H. C. Whitaker, I. G. Mills, M. Stewart and D. E. Neal, J. Cell Sci., 2008, 121, 957–968 CrossRef CAS.
- V. Chandra, P. Huang, Y. Hamuro, S. Raghuram, Y. Wang, T. P. Burris and F. Rastinejad, Nature, 2008, 456, 350–356 CrossRef.
- N. Rochel, F. Ciesielski, J. Godet, E. Moman, M. Roessle, C. Peluso-Iltis, M. Moulin, M. Haertlein, P. Callow, Y. Mely, D. I. Svergun and D. Moras, Nat. Struct. Mol. Biol., 2011, 18, 564–570 CAS.
- J. Zhang, M. J. Chalmers, K. R. Stayrook, L. L. Burris, Y. Wang, S. A. Busby, B. D. Pascal, R. D. Garcia-Ordonez, J. B. Bruning, M. A. Istrate, D. J. Kojetin, J. A. Dodge, T. P. Burris and P. R. Griffin, Nat. Struct. Mol. Biol., 2011, 18, 556–563 CAS.
- J. Reid, S. M. Kelly, K. Watt, N. C. Price and I. J. McEwan, J. Biol. Chem., 2002, 277, 20079–20086 CrossRef CAS.
- R. Kumar, R. Betney, J. Li, E. B. Thompson and I. J. McEwan, Biochemistry, 2004, 43, 3008–3013 CrossRef CAS.
- D. N. Lavery and I. J. McEwan, Biochemistry, 2008, 47, 3360–3369, DOI:10.1021/bi702221e.
- J. Brodie and I. J. McEwan, J. Mol. Endocrinol., 2005, 34, 603–615, DOI:10.1677/jme.1.01723.
- H. J. Dyson and P. E. Wright, Nat. Rev. Mol. Cell Biol., 2005, 6, 197–208, DOI:10.1038/nrm1589.
- P. Tompa, FEBS Lett., 2005, 579, 3346–3354, DOI:10.1016/j.febslet.2005.03.072.
- J. Liu, N. B. Perumal, C. J. Oldfield, E. W. Su, V. N. Uversky and A. K. Dunker, Biochemistry, 2006, 45, 6873–6888, DOI:10.1021/bi0602718.
- V. J. Hilser and E. B. Thompson, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 8311–8315, DOI:10.1073/pnas.0700329104.
- D. Gioeli, S. B. Ficarro, J. J. Kwiek, D. Aaronson, M. Hancock, A. D. Catling, F. M. White, R. E. Christian, R. E. Settlage, J. Shabanowitz, D. F. Hunt and M. J. Weber, J. Biol. Chem., 2002, 277, 29304–29314 CrossRef CAS.
- L. A. Ponguta, C. W. Gregory, F. S. French and E. M. Wilson, J. Biol. Chem., 2008, 283, 20989–21001 CrossRef CAS.
- S. S. Taneja, S. Ha, N. K. Swenson, H. Y. Huang, P. Lee, J. Melamed, E. Shapiro, M. J. Garabedian and S. K. Logan, J. Biol. Chem., 2005, 280, 40916–40924 CrossRef CAS.
- P. McCall, L. K. Gemmell, R. Mukherjee, J. M. Bartlett and J. Edwards, Br. J. Cancer, 2008, 98, 1094–1101 CrossRef CAS.
- M. Fu, C. Wang, A. T. Reutens, J. Wang, R. H. Angeletti, L. Siconolfi-Baez, V. Ogryzko, M. L. Avantaggiati and R. G. Pestell, J. Biol. Chem., 2000, 275, 20853–20860 CrossRef CAS.
- M. Fu, C. Wang, J. Wang, X. Zhang, T. Sakamaki, Y. G. Yeung, C. Chang, T. Hopp, S. A. Fuqua, E. Jaffray, R. T. Hay, J. J. Palvimo, O. A. Janne and R. G. Pestell, Mol. Cell. Biol., 2002, 22, 3373–3388 CrossRef CAS.
- L. Gaughan, I. R. Logan, S. Cook, D. E. Neal and C. N. Robson, J. Biol. Chem., 2002, 277, 25904–25913 CrossRef CAS.
- M. Fu, M. Rao, C. Wang, T. Sakamaki, J. Wang, D. Di Vizio, X. Zhang, C. Albanese, S. Balk, C. Chang, S. Fan, E. Rosen, J. J. Palvimo, O. A. Janne, S. Muratoglu, M. L. Avantaggiati and R. G. Pestell, Mol. Cell. Biol., 2003, 23, 8563–8575 CrossRef CAS.
- L. Gaughan, J. Stockley, N. Wang, S. R. McCracken, A. Treumann, K. Armstrong, F. Shaheen, K. Watt, I. J. McEwan, C. Wang, R. G. Pestell and C. N. Robson, Nucleic Acids Res., 2011, 39, 1266–1279 CrossRef CAS.
- S. Ko, J. Ahn, C. S. Song, S. Kim, K. Knapczyk-Stwora and B. Chatterjee, Mol. Endocrinol., 2011, 25, 433–444 CrossRef CAS.
- M. Fu, M. Rao, K. Wu, C. Wang, X. Zhang, M. Hessien, Y. G. Yeung, D. Gioeli, M. J. Weber and R. G. Pestell, J. Biol. Chem., 2004, 279, 29436–29449 CrossRef CAS.
- M. Thomas, N. Dadgar, A. Aphale, J. M. Harrell, R. Kunkel, W. B. Pratt and A. P. Lieberman, J. Biol. Chem., 2004, 279, 8389–8395 CrossRef CAS.
- Y. X. Chen, J. T. Du, L. X. Zhou, X. H. Liu, Y. F. Zhao, H. Nakanishi and Y. M. Li, Chem. Biol., 2006, 13, 937–944 CrossRef CAS.
- W. Chen, T. Dang, R. D. Blind, Z. Wang, C. N. Cavasotto, A. B. Hittelman, I. Rogatsky, S. K. Logan and M. J. Garabedian, Mol. Endocrinol., 2008, 22, 1754–1766 CrossRef CAS.
- A. M. Garza, S. H. Khan and R. Kumar, Mol. Cell. Biol., 2010, 30, 220–230 CrossRef CAS.
- P. Chymkowitch, N. Le May, P. Charneau, E. Compe and J. M. Egly, EMBO J., 2011, 30, 468–479 CrossRef CAS.
- H. Poukka, U. Karvonen, O. A. Janne and J. J. Palvimo, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 14145–14150 CrossRef CAS.
- L. Callewaert, G. Verrijdt, A. Haelens and F. Claessens, Mol. Endocrinol., 2004, 18, 1438–1449 CrossRef CAS.
- J. A. Iniguez-Lluhi and D. Pearce, Mol. Cell. Biol., 2000, 20, 6040–6050 CrossRef CAS.
- S. S. Dutt and A. C. Gao, Future Oncol., 2009, 5, 1403–1413 CrossRef CAS.
- C. D. Chen, D. S. Welsbie, C. Tran, S. H. Baek, R. Chen, R. Vessella, M. G. Rosenfeld and C. L. Sawyers, Nat. Med., 2004, 10, 33–39 CrossRef.
- J. Duff and I. J. McEwan, Mol. Endocrinol., 2005, 19, 2943–2954 CrossRef CAS.
- C. Tran, S. Ouk, N. J. Clegg, Y. Chen, P. A. Watson, V. Arora, J. Wongvipat, P. M. Smith-Jones, D. Yoo, A. Kwon, T. Wasielewska, D. Welsbie, C. D. Chen, C. S. Higano, T. M. Beer, D. T. Hung, H. I. Scher, M. E. Jung and C. L. Sawyers, Science, 2009, 324, 787–790 CrossRef CAS.
- H. C. Shen and S. P. Balk, Cancer Cell, 2009, 15, 461–463 CrossRef CAS.
- D. Roell and A. Baniahmad, Mol. Cell. Endocrinol., 2011, 332, 1–8 CrossRef CAS.
- I. A. Siddiqui, M. Asim, B. B. Hafeez, V. M. Adhami, R. S. Tarapore and H. Mukhtar, FASEB J., 2011, 25, 1198–1207 CrossRef CAS.
- J. O. Jones and M. I. Diamond, ACS Chem. Biol., 2008, 3, 412–418 CrossRef CAS.
- S. N. Quayle, N. R. Mawji, J. Wang and M. D. Sadar, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 1331–1336 CrossRef CAS.
- M. D. Sadar, D. E. Williams, N. R. Mawji, B. O. Patrick, T. Wikanta, E. Chasanah, H. E. Irianto, R. V. Soest and R. J. Andersen, Org. Lett., 2008, 10, 4947–4950 CrossRef CAS.
- R. J. Andersen, N. R. Mawji, J. Wang, G. Wang, S. Haile, J. K. Myung, K. Watt, T. Tam, Y. C. Yang, C. A. Banuelos, D. E. Williams, I. J. McEwan, Y. Wang and M. D. Sadar, Cancer Cell, 2010, 17, 535–546 CrossRef CAS.
- P. A. Watson, Y. F. Chen, M. D. Balbas, J. Wongvipat, N. D. Socci, A. Viale, K. Kim and C. L. Sawyers, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 16759–16765 CrossRef CAS.
- S. Sun, C. C. Sprenger, R. L. Vessella, K. Haugk, K. Soriano, E. A. Mostaghel, S. T. Page, I. M. Coleman, H. M. Nguyen, H. Sun, P. S. Nelson and S. R. Plymate, J. Clin. Invest., 2010, 120, 2715–2730 CAS.
- R. Hu, W. B. Isaacs and J. Luo, Prostate, 2011 DOI:10.1002/pros.21382; 10.1002/pros.21382.
- E. Hornberg, E. B. Ylitalo, S. Crnalic, H. Antti, P. Stattin, A. Widmark, A. Bergh and P. Wikstrom, PLoS One, 2011, 6, e19059 Search PubMed.
- S. M. Dehm, L. J. Schmidt, H. V. Heemers, R. L. Vessella and D. J. Tindall, Cancer Res., 2008, 68, 5469–5477 CrossRef CAS.
- L. Ni, C. S. Yang, D. Gioeli, H. Frierson, D. O. Toft and B. M. Paschal, Mol. Cell. Biol., 2010, 30, 1243–1253 CrossRef CAS.
- C. W. Kuil and E. Mulder, Mol. Cell. Endocrinol., 1994, 102, R1–5 CrossRef CAS.
- D. J. Osguthorpe and A. T. Hagler, Biochemistry, 2011, 50, 4105–4113 CrossRef CAS.
- M. Papaioannou, S. Schleich, D. Roell, U. Schubert, T. Tanner, F. Claessens, R. Matusch and A. Baniahmad, Invest. New Drugs, 2010, 28, 729–743 CrossRef CAS.
- N. G. Nickols and P. B. Dervan, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 10418–10423 CrossRef CAS.
- J. O. Jones, E. C. Bolton, Y. Huang, C. Feau, R. K. Guy, K. R. Yamamoto, B. Hann and M. I. Diamond, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 7233–7238 CrossRef CAS.
Footnote |
| † Published as part of a Molecular BioSystems themed issue on Intrinsically Disordered Proteins: Guest Editor M Madan Babu. |
| This journal is © The Royal Society of Chemistry 2012 |