Structural disorder within paramyxovirus nucleoproteins and phosphoproteins†
Johnny
Habchi
and
Sonia
Longhi
*
Architecture et Fonction des Macromolécules Biologiques, UMR 6098 CNRS et Universités d'Aix-Marseille I et II, 163, Avenue de Luminy, Case 932, 13288 Marseille Cedex 09, France. E-mail: Sonia.Longhi@afmb.univ-mrs.fr; Fax: +33 4 91 26 67 20; Tel: +33 4 91 82 55 80
First published on 1st August 2011
Abstract
This review focuses on the experimental data showing the abundance of structural disorder within the nucleoprotein (N) and phosphoprotein (P) from three paramyxoviruses, namely Nipah (NiV), Hendra (HeV) and measles (MeV) viruses. We provide a detailed description of the molecular mechanisms governing the disorder-to-order transition of the intrinsically disordered C-terminal domains (NTAIL) of their N proteins upon binding to the C-terminal X domain (XD) of the homologous P proteins. We also show that a significant flexibility persists within NTAIL–XD complexes, which therefore provide illustrative examples of “fuzziness”. The functional implications of structural disorder are discussed in light of the ability of disordered regions to establish a complex molecular partnership, thereby leading to a variety of biological effects. Taking into account the promiscuity that typifies disordered regions, we propose that the main functional advantage of the abundance of disorder within viruses would reside in pleiotropy and genetic compaction, where a single gene would encode a single (regulatory) protein product able to establish multiple interactions via its disordered regions, and hence to exert multiple concomitant biological effects.
 Johnny Habchi | Johnny Habchi is a PhD student within the “Structural disorder and Molecular Recognition” team at the AFMB laboratory under the supervision of Dr Sonia Longhi. After obtaining the bachelor degree from the “Lebanese University of Beirut” in 2008, he achieved his Masters studies in “Structural Biology” in 2009 at the “Université de la Méditérranée” (Aix-Marseille II). So far, he has authored four papers with two of them focusing on the characterization of the intrinsically disordered regions within the replicative complex of Henipaviruses and the induced folding that these regions undergo in the presence of their partners. |
 Sonia Longhi | Sonia Longhi is Director of Research at the Center for the National Scientific Research (CNRS), heading the “Structural Disorder and Molecular Recognition” group within the AFMB laboratory. She obtained a PhD in molecular biology from the Universitá degli Studi of Milan in 1993. She got a HDR in structural virology from the University of Aix-Marseille I in 2003. Her scientific focus is on intrinsically disordered proteins (IDPs) and the mechanistic and functional aspects of the interactions they establish with partners. She has authored more than sixty scientific publications, edited a book on measles virus nucleoprotein and co-edited (with Prof. Vladimir Uversky) a book on the experimental approaches to characterize IDPs and the folding coupled to binding events they undergo in the presence of partners. |
1. The replicative complex of measles, Nipah and Hendra viruses
Nipah (NiV), Hendra (HeV) and measles (MeV) viruses belong to the Paramyxoviridae family within the Mononegavirales order.1–3 While MeV belongs to the Morbillivirus genus, NiV and HeV have been classified within the recently emerged Henipavirus genus. Paramyxoviruses are enveloped viruses possessing a non-segmented, single-stranded negative sense RNA genome, which encodes six proteins, namely the nucleoprotein (N), the phosphoprotein (P), the matrix protein (M), the F and H glycoproteins and the polymerase or “large” (L) protein. As in all Mononegavirales members, the genome is encapsidated by N to form a helical nucleocapsid. The N![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ∶RNA complex, rather than naked RNA, is the template for both transcription and replication. These latter activities are carried out by the RNA dependent RNA polymerase that is composed of L and P. The P protein is an essential polymerase co-factor in that it tethers the L protein onto the nucleocapsid template. This ribonucleoprotein complex made of RNA, N, P and L constitutes the basic elements of the viral transcriptase and replicase (i.e., the viral replicative unit) (Fig. 1).
∶RNA complex, rather than naked RNA, is the template for both transcription and replication. These latter activities are carried out by the RNA dependent RNA polymerase that is composed of L and P. The P protein is an essential polymerase co-factor in that it tethers the L protein onto the nucleocapsid template. This ribonucleoprotein complex made of RNA, N, P and L constitutes the basic elements of the viral transcriptase and replicase (i.e., the viral replicative unit) (Fig. 1).
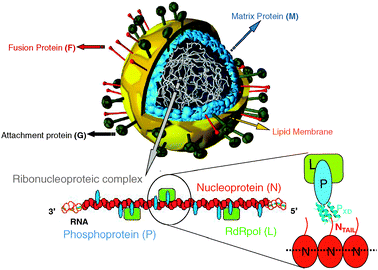 | ||
| Fig. 1 Schematic illustration of a Paramyxovirus particule. The virion is enveloped by a lipid bilayer membrane in which are inserted the glycoproteins. Beneath the envelop, the matrix protein associates with the cytoplasmic tails of the H and F proteins as well as with the ribonucleoproteic complex. This latter, made of the viral RNA, and of the N, P and L proteins, is shown below the viral particle. The encapsidated RNA is shown as a green dotted line embedded in the middle of N by analogy with RV, VSV and RSV N∶RNA complexes.93,94,96 The L protein is tethered onto the nucleocapsid by its cofactor, the P protein. Binding of N to P is mediated by the interaction between their NTAIL and X domains. Modified from www.uct.ac.za/Depts/MMI/Stannard/syncytia.html (Linda Stannard, Department of Medical Microbiology, University of Capetown, South Africa). | ||
By virtue of their structural role in encapsidating the genome, the nucleoproteins from MeV, NiV and HeV are the most abundant viral proteins. Within MeV infected cells, N is found in a soluble, monomeric form (referred to as N°) and in a nucleocapsid assembled form. Following synthesis of the N protein, a chaperone is required to maintain this latter protein in a soluble and monomeric form. This role is played by the P protein, whose association simultaneously prevents illegitimate self-assembly of N.4,5 This soluble N°–P complex is used as the substrate for the encapsidation of the nascent genomic RNA chain during replication (see ref. 1, 6–9 for reviews on transcription and replication). The assembled form of N also forms complexes with either isolated P or P bound to L, which are both essential to RNA synthesis by the viral polymerase.10,11
The viral polymerase, which is responsible for both transcription and replication, is poorly characterized. It is thought to carry out most (if not all) enzymatic activities required for transcription and replication, including nucleotide polymerization, mRNA capping and polyadenylation. So far no functional Paramyxoviridae polymerase has been purified to homogeneity, with the only exception of the Rinderpest virus L–P complex, which has been partially purified.12 A methyltransferase activity has been demonstrated biochemically within the C-terminal region of the closely related Sendai virus (SeV, a respirovirus) polymerase (aa 1756–2228),13 consistent with previous bioinformatics analysis.14 Interestingly, both ribose-2′-O and guanosine–N-7 methyltransferase activities have been mapped to a conserved C-terminal motif of the L protein from the vesicular stomatitis virus (VSV, a Rhabdoviridae member)15 using a purified recombinant form of L.16
Although the understanding of the precise role(s) of N, P and L within the replicative complex of MeV has benefited of significant breakthroughs in recent years (see ref. 17–20 for reviews), rather limited three-dimensional information on the replicative machinery is available. In the case of Henipaviruses, even less structural data are available, with only one paper focused on the structural characterization of their N and P proteins having been published so far.21 As for the N–P interaction, the only available data come from studies carried out by Chan et al.22 and from our recently published studies.23
The paucity of high-resolution structural data on N and P proteins arises from the difficulty of obtaining homogenous polymers of N suitable for X-ray analysis and from the structural flexibility of N and P. Indeed, in the course of the structural and functional characterization of MeV, NiV and HeV N and P proteins, we discovered that they contain long (up to 400 residues) disordered regions that possess the sequence and biochemical features that typify intrinsically disordered proteins (IDPs).17,19,21,24–29 IDPs are functional proteins that lack highly populated secondary and tertiary structure under physiological conditions of pH and salinity in the absence of a partner, and exist as dynamic ensembles of conformers.30–34
Using various computational approaches (as described in),35,36 we further extended these results to the N and P proteins of viruses of the Paramyxovirinae subfamily.26 By combining computational and experimental approaches (as described in),37,38 we showed that large disordered regions also occur within the P protein of rabies virus (RV, a Rhabdoviridae member)39 and of respiratory syncytial virus (RSV, a Pneumovirinae member within the Paramyxoviridae family).40 Altogether these data pointed out that structural disorder is a conserved and widespread property within these two viral families, thus implying functional relevance.
2. Modular organization and structural disorder within MeV, NiV and HeV P proteins
Beyond serving as a chaperone for N, P binds to the nucleocapsid, thus tethering the polymerase onto the nucleocapsid template. The actual oligomeric state of MeV and Henipavirus P is unknown. However, by analogy with the closely related SeV,41,42 they are thought to be tetrameric.The P genes of MeV, NiV and HeV encode multiple proteins, including P, V and C (for reviews see ref. 1, 3, 6 and 7). While the C protein is encoded by an alternate ORF within the P gene through ribosome initiation at an alternative translation codon, the V protein is translated from a P messenger obtained upon co-transcriptional insertion of a G at the editing site of the P mRNA. The V protein thus shares with the P protein the N-terminal module (MeV PNT, aa 1–230; NiV PNT, aa 1–406 and HeV PNT, aa 1–404) and possesses a unique C-terminal, zinc binding domain. Hence, the modular organization of the P protein consists of at least two domains: an N-terminal domain (PNT) common to both P and V, and a C-terminal domain (PCT) unique to the P protein (see Fig. 2).
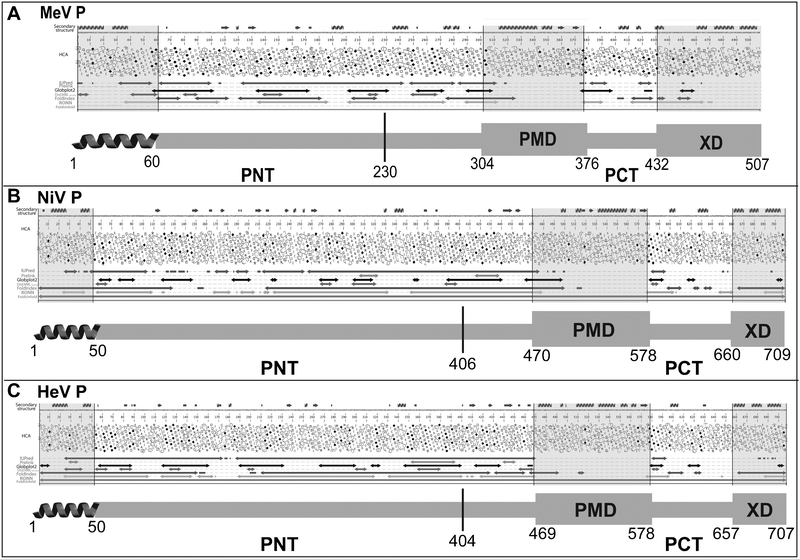 | ||
| Fig. 2 Disorder prediction and modular organization of the P proteins from MeV (A), NiV (B) and HeV (C). The output of MeDor and the inferred modular organization of P are shown on the top and bottom of each panel, respectively. The sequences are represented as single, continuous horizontal lines below the predicted secondary structure elements. Below the sequences are shown the HCA plots and the predicted regions of disorder that are represented by bi-directional arrows. Regions shaded in grey within the MeDOr output correspond to either putative N-terminal α-MoREs or structured regions (PMD and XD) within PCT. Structured and disordered regions are represented by large and narrow boxes, respectively. The vertical line separating PNT and PCT is located at the border between the region shared by the P and V proteins, and the region unique to P (see text). | ||
The PNT domains from MeV, NiV and HeV are consistently predicted to be disordered by several disorder predictors,21,24 including the method of the hydrophobicity/mean charge ratio,43 as well as most disorder predictors implemented within the MeDor metaserver for the prediction of disorder.44 Indeed, as much as 90% of the PNT domains is depleted in hydrophobic clusters and possesses very few predicted secondary structure elements, a feature typifying protein regions with “no ordered regular structure” (see Fig. 2). The disordered state of NiV, HeV and MeV PNT domains was experimentally assessed using complementary biochemical and biophysical approaches.21,24 The hydrodynamic properties of PNT domains inferred from size exclusion chromatography (SEC) are consistent with these protein domains possessing extended conformations in solution. Moreover, the absence of a globular core has been further demonstrated by limited proteolysis experiments, which showed that PNT domains are fully exposed to the solvent. In addition, PNT domains display NOESY and far-UV circular dichroism (CD) spectra typical of unfolded proteins.21,24
Interestingly, a short (40–50 residues) ordered region is consistently predicted at the N-terminus of MeV, NiV and HeV P by all the predictors implemented in MeDor (Fig. 2). This N-terminal module with α-helical folding propensities corresponds to a conserved region amongst Avulavirus, Henipavirus and Rubulavirus members,26 with that from Rubulaviruses having been shown to be involved in N°-binding.45 A similar N-terminal module globally disordered yet containing transient α-helices (aa 1–60) has recently been identified and characterized in the VSV P protein,46 and previously shown to interact with N°.47 This region likely corresponds to an α-helical Molecular Recognition Element (α-MoRE), where MoREs are short, order-prone regions within IDPs that have a certain propensity to bind to a partner and thereby to undergo induced folding (i.e. a disorder-to-order transition).48–51 In agreement with the predicted occurrence of a transiently populated α-MoRE, SEC and dynamic light scattering (DLS) studies indeed unveiled that MeV, NiV and HeV PNT domains are not fully unfolded and rather conserve some degree of compactness typical of a premolten globule (PMG) conformation.21,24 PMG are characterized by a conformational state intermediate between a random coil and a molten globule and possess a certain degree of residual compactness due to the presence of residual and fluctuating secondary and/or tertiary structures.52,53 The folding potential of the PNT domains was further confirmed using far-UV CD spectroscopy, where increasing concentrations of 2,2,2-trifluoroethanol (TFE), were shown to induce a pronounced gain of α-helicity within these domains.21,24TFE is an organic solvent that mimics the hydrophobic environment experienced by proteins during protein–protein interactions.54,55 The extent of residual compactness within the three PNT domains follows the order NiV PNT > HeV PNT > MeV PNT. It has been proposed that the residual intramolecular interactions that typify the PMG state may enable a more efficient start of the folding process induced by a partner by lowering the entropic cost of the folding-coupled-to-binding process.56–58
Beyond PNT, other disordered regions have been identified within MeV and Henipavirus P. Indeed, PCT has a modular organization, being composed of alternating disordered and structured regions21,26 (see Fig. 2). In Morbilliviruses and Henipaviruses, sequence analysis predicts a coiled-coil region within the P multimerization domain (PMD), which is the region responsible for the oligomerization of P.59 The coiled-coil organization has been experimentally confirmed in the case of SeV42 and Rinderpest virus60 PMDs, whereas RV61 and VSV62 PMDs, were shown to form dimers with a different structural arrangement.
Computational analyses predict three α-helices within the C-terminal X domains (XD, aa 459–507 for MeV, aa 660–709 for NiV and 657–707 for HeV) of P21,26 (see Fig. 2). In agreement with these predictions, the crystal structure of MeV XD was shown to consist of a triple α-helical bundle,63 and CD and NMR studies of Henipavirus X domains showed that they are folded and adopt a predominantly α-helical conformation.64 Strikingly, in spite of the high similarities between Henipavirus X domains (about 90% of similarity and 80% of identity), NiV XD was found to be trimeric while HeV XD was found to be monomeric in size exclusion chromatography coupled to multiple angle laser light scattering (SEC-MALLS) studies.64 In all the three paramyxoviruses, XD was shown to be the P region responsible for binding to the C-terminal region of N.63–65
High-resolution structural data are also available for the X domains of the closely related SeV and mumps virus (MuV), the structure of which have been solved by nuclear magnetic resonance (NMR) and X-ray crystallography, respectively.66,67 The MuV X domain (aa 343–391 of P) has a few notable distinguishing properties with respect to MeV and SeV XD. Indeed, MuV XD has been shown to exist as a molten globule in solution, being loosely packed and devoid of stable tertiary structure.66,68 In addition, contrary to the MeV and SeV X domains, MuV XD does not interact with the C-terminal region of N and rather establish contacts with the structured NCORE region of N.68 Structural data are also available for the C-terminal P domains from Rhabdoviridae members, namely RV,69VSV70 and Mokola virus.71 Interestingly, comparison of the P nucleocapsid-binding domains solved so far suggests that the nucleocapsid binding domains are structurally conserved among Paramyxoviridae and Rhabdoviridae P in spite of low sequence conservation thus providing a fascinating example of convergent evolution.72
In all Paramyxovirinae, PMD and XD are separated by a flexible linker region predicted to be poorly ordered.26 Indeed, in the case of SeV, NMR studies carried out on the 474–568 region of P (referred to as PX) showed that the region upstream XD (aa 474–515 of P) is disordered.73,74 In Morbilliviruses and Henipaviruses, an additional flexible region (referred to as “spacer”) is predicted to occur upstream PMD.21,26
3. Modular organization and structural disorder within MeV, NiV and HeV N proteins
Bioinformatics, deletion and electron microscopy studies have shown that Paramyxoviridae nucleoproteins are divided into two regions: a structured N-terminal moiety, NCORE (aa 1–400 in MeV and aa 1–399 in Henipaviruses), which contains all the regions necessary for self-assembly and RNA-binding,68,75–81 and a C-terminal domain, referred to as NTAIL (aa 401–525 in MeV and aa 400–532 in Henipaviruses). Altogether, the NTAIL domains from MeV, NiV and HeV possess features that are hallmarks of intrinsic disorder: (i) they are hyper-sensitive to proteolysis,23,81 (ii) they cannot be visualized in cryo-electron microscopy reconstructions of nucleocapsids,82 (iii) they have an amino acid sequence that is highly variable amongst phylogenetically related members21 and (iv) they have a significant sequence compositional bias21,25,27 being enriched in disorder-promoting residues and depleted in order-promoting residues.53 In agreement, they are predicted to be mainly (if not fully) disordered by the secondary structure and disorder predictors implemented within the MeDor metaserver44 (see Fig. 3). The disordered nature of these NTAIL domains has been further confirmed experimentally by both hydrodynamic and spectroscopic approaches, which showed that they all belong to the PMG subfamily.21,25,27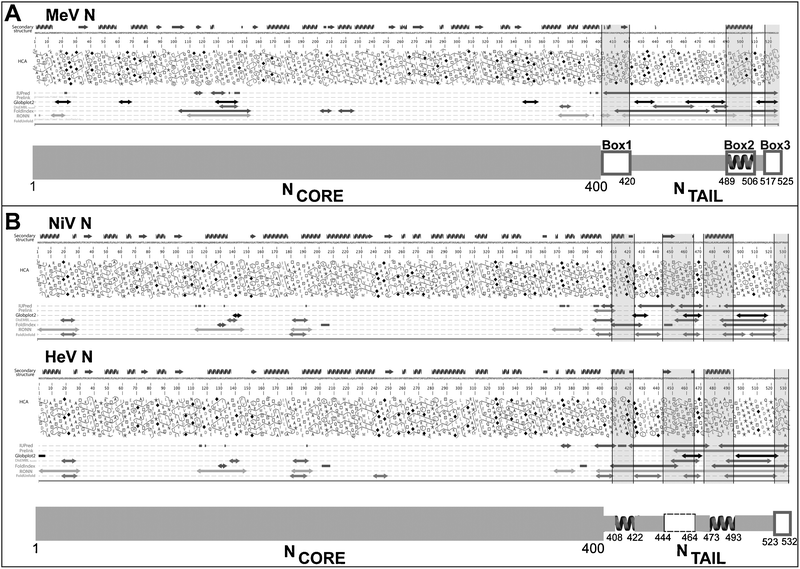 | ||
| Fig. 3 Disorder prediction and modular organization of the N proteins from MeV (A) and Henipaviruses (B). The output of MeDor and the inferred modular organization of N are shown on the top and bottom of each panel, respectively. The sequences are represented as single, continuous horizontal lines below the predicted secondary structure elements. Below the sequences are shown the HCA plots and the predicted regions of disorder that are represented by bi-directional arrows. Regions shaded in grey within the MeDOr output correspond to putative MoREs within NTAIL. Structured and disordered regions are represented by large and narrow boxes, respectively. (A) The three NTAIL boxes—namely Box1 (aa 400–420), Box2 (aa 489–506) and Box3 (aa 517–525)—that are conserved among Morbillivirus members97 are shown. The experimentally characterized α-MoRE involved in binding to XD within Box2 is shown.65 (B) Two putative α-MoREs (aa 408–422 and aa 473–493) with the second one corresponding to the well-characterized α-MoRE of MeV-NTAIL, are shown. A putative irregular MoRE (I-MoRE) (aa 523–532) and a putative MoRE of dubious state (aa 444–464, see region contoured by a dashed line) are also shown. | ||
Like for all N proteins of Mononegavirales members, and with the only exception of the N protein from the Borna disease virus (BDV),83MeV and Henipavirus N proteins self-assemble to form large helical nucleocapsid-like particles with a broad size distribution when expressed in heterologous systems84–88 (Papageorgiou, Habchi and Longhi, unpublished data). MeV nucleocapsids, as visualized by negative stain transmission electron microscopy, have a typical herringbone-like appearance.25,81,82,86,89Electron microscopy studies by two independent groups led to real-space helical reconstruction of MeV nucleocapsids.82,89 These studies showed that the removal of the disordered NTAIL domain, which protrudes from the globular body of NCORE and is at least partly exposed at the surface of the viral nucleocapsid,81,90,91 leads to increased nucleocapsid rigidity, with significant changes in both pitch and twist (see).25,82,89,92
So far, relatively few high-resolution structural data on N are available. In contrast with BDV N, the structure of which has been solved in an RNA-free form,83 the structures of RV and VSV,93–95 and of RSV96 were crystallized in the form of N∶RNA rings. The nucleoprotein of these latter viruses consists of two lobes and possesses an extended terminal arm that makes contacts with a neighboring N monomer. The RNA is tightly packed between the two N lobes and, in the case of RV and VSV, points towards the inner cavity of the N∶RNA rings. Conversely, in the case of RSV, the RNA is located on the external face of the N∶RNA rings.96 Using the structure of RSV N∶RNA rings as a template, a model of MeV N∶RNA has been recently built and docked within the electron density map of MeV nucleocapsids.92 Although the disordered NTAIL domain could not be resolved in the reconstruction of the nucleocapsid, the fit suggests that while the RNA is located at the exterior of the helical nucleocapsid, NTAIL would point toward the helix interior.92 Thus, in all these nucleoproteins, the RNA is not accessible to the solvent, and has to be partially released from N to become accessible to the polymerase. Therefore, a conformational change must occur within N to allow exposure of the RNA. The disordered NTAIL domain is thought to play a major role in this conformational change.
Although HeV, NiV and MeV NTAIL domains are mostly disordered, bioinformatics and biochemical analyses pointed out the presence of short order-prone regions, possibly corresponding to MoREs (see Fig. 3). In the case of MeV, an α-MoRE involved in binding to XD and in the α-helical folding of NTAIL has been identified27 within one (namely Box2, aa 489–506 of N) out of three NTAIL regions (referred to as Box1, Box2 and Box3) that are conserved within Morbillivirus members97 (see Fig. 3A). Beyond this α-MoRE, two additional short regions with some folding potential can be detected within MeV NTAIL: one located in the 400–420 region and one located at the C-terminus (aa 516–525) (see Fig. 3A). Notably, the former was shown to represent the binding site to a yet uncharacterized Nucleoprotein Receptor, referred to as NR, which is expressed at the surface of dendritic cells of lymphoid origin (both normal and tumoral),98 and of T and B lymphocytes,99 while the latter, has been shown to play a role in the binding to XD (see Section 4).
In the case of HenipavirusNTAIL, four putative MoREs were identified (see Fig. 3B).21 The one encompassing residues 473–493 roughly corresponds to the above-mentioned experimentally characterized α-MoRE of MeV NTAIL,27,63,65 and has hence been proposed to constitute the XD-binding site of HenipavirusNTAIL.23
Beyond being disordered in isolation, the NiV, HeV and MeV NTAIL domains were also shown to be disordered within full-length N proteins from nucleocapsid-like particles, as testified by the observation that the N proteins of these viruses undergo proteolytic cleavage within their NTAIL domains.64,81 In the same vein, recently Ringkjøbing Jensen et al. reported the first in situ structural characterization of MeV NTAIL in the context of the entire N-RNA nucleocapsid.100 Using NMR and small angle scattering, they showed that NTAIL is highly flexible in intact nucleocapsids. Moreover, that study also provided experimental evidence supporting a model in which the first 50 disordered amino acids of NTAIL are conformationally restricted as the chain escapes to the outside of the nucleocapsidvia the interstitial space between successive NCORE helical turns.100 Notably, this model provides a plausible explanation for the increased rigidity of nucleocapsids in which the flexible NTAIL region has been cleaved off. The inherent flexibility of intact nucleocapsids likely confers at least partial accessibility to the N-terminal region of NTAIL, thereby accounting for the ability of the Box1 region to bind to NR in the context of nucleocapsids released in the extracellular compartment.98,99 This same flexibility probably also accounts for the ability of trypsin to cleave off NTAIL in (limited) proteolysis studies, where the major cleavage site has been located at residue 392.81
4. Molecular mechanisms of NTAIL–XD complex formation
The NTAIL domains of MeV, NiV and HeV were shown to undergo induced folding upon binding to the C-terminal X domain of the homologous P protein.23,63 In the case of MeV, a model of the interaction in which the α-MoRE of NTAIL adopts an α-helical conformation and is embedded in a large hydrophobic cleft delimited by helices α1 and α2 of XD has been proposed63 and successively validated65 (see Fig. 4A).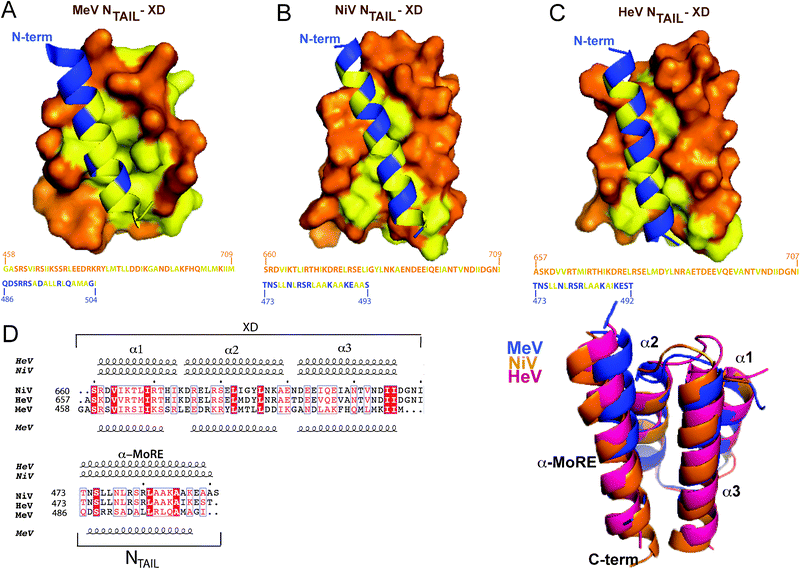 | ||
| Fig. 4 Structure of NTAIL–XD complexes. (A) Structure of the MeV chimeric NTAIL–XD construct (PDB code 1T6O)65 in which MeV XD (amino acids 459–507 of P) is shown in orange with surface presentation and the α-MoRE of NTAIL (residues 486–504 of N) is shown in blue with ribbon representation. Hydrophobic residues are shown in yellow. The amino acid sequence of XD and of the α-MoRE are shown with the same color code. Structural models of the NiV (B) and HeV (C) NTAIL–XD complexes, in which the NTAIL region predicted to adopt an α-helical conformation (amino acids 473–493 for NiV and 473–492 for HeV) is shown in blue with ribbon representation, while XD is shown in orange with surface representation. Hydrophobic residues are shown in yellow. The amino acid sequences of XD and of the α-MoRE are shown with the same color code. (D) Superimposition of the structural models (ribbon representation) of the NTAIL–XD complexes of HeV (pink) and NiV (orange) onto the crystal structure of the MeV chimeric construct (blue). A multiple sequence alignment of Henipavirus and MeV NTAIL and XD as obtained using ClustalW156 and ESPript157 is also shown. Residues corresponding to a similarity greater than 60% are boxed and shown in red. Identical residues are boxed and shown in white on a red background. The numbers written in front of the sequences correspond to the amino acid positions in the P and N sequences. Dots above the alignment indicate intervals of 10 residues. Predicted secondary structure elements, as obtained using the PSIPRED server,158 are shown above the multiple sequence alignment. Secondary structure elements, as observed in the crystal structure of the MeV chimeric construct (PDB code 1T6O) are shown below the alignment. All structural models were drawn using PyMOL.159 Modified from ref. 23. | ||
Although NiV XD is trimeric, it binds to NTAIL with a 1∶1 stoichiometry64 as in the case of MeV63,68 and HeV.64 While HenipavirusNTAIL–XD complexes are characterized by an equilibrium dissociation constant (KD) in the μM range (2 μM for NiV and 8 μM for HeV), as judged from isothermal titration calorimetry (ITC) and surface plasmon resonance (SPR) studies,23 the affinity of the MeV NTAIL–XD complex is in the nM range (100 nM).28
In the case of the MeV NTAIL–XD complex, SPR studies showed that the removal of Box3 (aa 518–525 of N) alone or of Box2 plus Box3 (aa 489–525 of N) results in a strong increase (three orders of magnitude) in the KD, which increases from 100 nM to either 12 μM (NTAILΔ3) or 41 μM (NTAILΔ2,3). The residual binding capacity of the truncated form devoid of Box2 plus Box3 likely results from the presence in the construct of residues 475–488, which have been shown to be significantly, though less dramatically, affected by the addition of XD, with residues 475–482 being in fast exchange.101
These results, together with the lack of a protruding appendage corresponding to the C-terminus of NTAIL in a low-resolution small angle X-ray scattering (SAXS) model of the MeV NTAIL–XD complex,28 suggest that beyond Box2, Box3 may also be involved in binding to XD. That XD does affect the Box3 region of MeV NTAIL was further confirmed by fluorescence spectroscopy studies showing a dose-dependent effect of XD on the fluorescence intensity of an NTAIL variant bearing a tyr to trp substitution within Box3 (position 518),28 as well as by heteronuclear NMR (HN-NMR) studies that showed that addition of XD triggers a significant chemical shift variation within Box3 (although much less pronounced than that observed within Box2).101
Surprisingly however, SPR and HN-NMR studies using either truncated NTAIL proteins or synthetic peptides mimicking Box1, Box2 and Box3 showed lack of significant affinity between XD and a Box3 peptide, suggesting that Box3 would act only in the context of NTAIL and not in isolation, being functionally coupled to Box2 in the binding of NTAIL to XD.20,102
In striking contrast with these data, in a recent study by the group of Kingston, where synthetic peptides corresponding to the 477–505 or 477–525 region of NTAIL were used, no evidence was obtained in support of a role of the 505–525 region in binding to XD.103 In addition, in that study, the experimentally determinedKD of the XD binding reaction was found to be either 7.4 μM or 15 μM depending on whether the 477–525 or the 477–505 peptide was used, respectively.103
A KD in the μM range, as the one observed upon removal of Box3 for the MeV NTAIL–XD couple, is similar to the KD observed for HenipavirusNTAIL–XD complexes.23 Strikingly, in these latter complexes, the C-terminal region of both NiV and HeV NTAIL domains was shown not to be involved in binding to XD, as judged from intrinsic fluorescence spectroscopy studies23 similar to those carried out on MeV NTAIL.28 Interestingly, in the case of the SeV NTAIL–XD complex, the KD is also in the μM range (60 μM) with no evidence for an involvement of the C-terminal region in NTAIL–XD complex formation.104 It is therefore tempting to speculate that the higher stability of the MeV complex with respect to that of the Henipavirus and SeV complexes could be correlated with a Box3 contribution to binding: in the MeV NTAIL–XD complex, Box3 would be less flexible than in the cognate complexes and would therefore contribute to further stabilize the bound conformation thereby leading to a higher affinity.
ITC studies revealed that HenipavirusNTAIL–XD complexes are stable under NaCl concentrations as high as 1 M suggesting that the interaction does not rely on polar contacts.23 That makes perfect sense for an interaction driven by the burying of apolar residues of NTAIL at the XD surface, as already observed in the case of MeV.63,68 The hydrophobic nature of the NTAIL–XD interface is in agreement with the findings by Meszaros and co-workers who reported that the binding interfaces of protein complexes involving IDPs are often enriched in hydrophobic residues.105 We therefore modeled the more hydrophobic side of the amphipathic α-MoRE of HenipavirusNTAIL at the hydrophobic surface delimited by helices α2 and α3 of XD using the MeV NTAIL–XD structure as a template (Fig. 4A). The resulting models (Fig. 4B and C) well superimpose on the MeV NTAIL–XD structure (Fig. 4D) and consist of a pseudo-four-helix arrangement that occurs frequently in nature.23 The two modeled complexes display a rather small interface area (439 Å2 for NiV and 337 Å2 for HeV) in agreement with previous reports indicating that the interfaces of complexes involving IDPs are generally smaller than those occurring in ordered complexes.51 The lower buried surface area of the HenipavirusNTAIL–XD complex as compared to the MeV NTAIL–XD couple (634 Å2) is consistent with the lower affinity of the binding reaction. Strikingly, charged residues dominate the SeV NTAIL–XD interface, which gives a good illustration of how selection pressure allowed the C-terminal domains of N and P to evolve concomitantly within the paramyxovirus family in order to lead to protein complexes having the same 3D fold and the same function, but with very limited sequence identity.104
Far-UV CD spectroscopy revealed that binding of HenipavirusNTAIL domains to XD results in the same type of structural transition (i.e. α-helical) observed with the MeV NTAIL–XD couple.28,63,64 Consistently, NMR titration experiments with 15N-labeled NTAIL, pointed out an α-helical transition within MeV and NiV NTAIL upon addition of XD, as judged from the appearance of new peaks in the α-helical region of the NTAIL spectra.23,28,101 Interestingly, in the case of HeV, no such peaks were observed in the NTAIL spectrum even with saturating amounts of XD and a few peaks were found to disappear at the beginning of titration and to never come back even at saturation. This behavior, which is often observed for IDPs undergoing folding-upon-binding events,106–108 supports an intermediate exchange regime among an ensemble of NTAIL conformers at the XD surface, thus arguing for a considerable conformational heterogeneity in the bound form (see Section 5).23 In all cases, the majority of NTAIL peaks were unaffected upon addition of XD, with affected residues being displaced according to a fast to intermediate regime.23,28,101 Definite answers about the HenipavirusNTAIL residues that are involved in the interaction with XD await the assignment of the NMR spectrum of the free and bound forms of NTAIL domains, a work that is currently in progress.
In the case of MeV NTAIL, the availability of the full spectral assignment of the free and bound form,101 together with the hints provided by site-directed spin labeling (SDSL) electron paramagnetic resonance (EPR) spectroscopy studies,109–111 greatly contributed to unravel the molecular mechanisms of the NTAIL–XD complex formation. In these latter studies, 14 single-site MeV NTAIL cysteine variants were designed, purified and labeled and their EPR spectra were recorded in the presence or absence of either XD or 20% TFE.109,110 The mobility of the spin labels grafted within the 488–502 (i.e. Box2) and 505–522 regions was found to be severely and moderately reduced, respectively, upon addition of XD.110 The restrained motion of the 505–522 region upon binding to XD was shown to be due to the α-helical transition occurring within the neighboring Box2 region and not to a direct interaction with XD. It has been proposed that the reduced conformational freedom of Box3 resulting from binding to XD may favor the establishment of weak, non-specific contacts with this latter, possibly stabilizing the bound form. The mobility of the 488–502 region was found to be restrained even in the absence of the partner, a behavior that could be accounted for by the existence of a transiently populated folded state. That the Box2 region is at least partly pre-structured prior to binding to XD has been confirmed by the analysis of the Cα chemical shifts of the free form,101 as well as by recent studies carried out by Ringkjøbing Jensen et al.100 In those studies, an atomic resolution ensemble description of isolated MeV NTAIL has been achieved using recently developed tools designed to provide quantitative descriptions of conformational equilibria in IDPs on the basis of experimental NMR data.73,112 Chemical shifts and residual dipolar couplings RDCs,73,112 were combined to directly probe the level and nature of residual structure in MeV NTAIL, revealing that while the majority of NTAIL behaves like an intrinsically disordered chain, the α-MoRE exists in a rapidly interconverting conformational equilibrium between an unfolded form and conformers containing four discrete α-helical elements situated around the interaction site (Fig. 5A). All of these α-helices are stabilized by N-capping interactions mediated by side chains of four different aspartic acids or serines that precede the observed helices.113 N-capping stabilization of helices or turns represents an important mechanism by which the primary sequence encodes pre-recognition states, and has already been observed in other IDPs including SeV NTAIL.112 Noteworthy, in the crystal structure of a chimeric MeV NTAIL–XD complex made of XD and of residues 486–504 of NTAIL, NTAIL adopts an α-helical conformation between residues Q486 and A502.65 This helix is similar to the longest of the four helical elements present in isolated NTAIL. Changes in chemical shifts and RDCs confirm that upon binding to XD, the α-MoRE folds into a helix.100,101
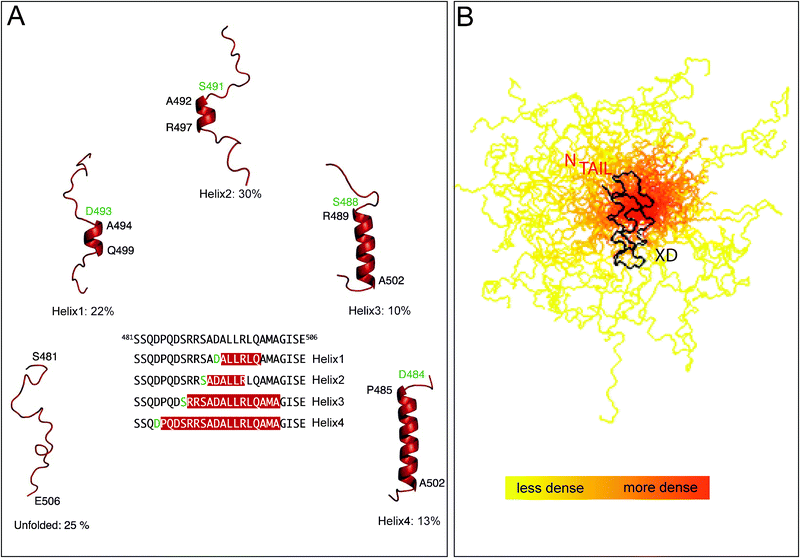 | ||
| Fig. 5 (A) Ensemble description of the MeV NTAIL α-MoRE. Four conformational states of the free form of MeV NTAIL, as derived by NMR spectroscopy, are represented by cartoon structures. All helices are stabilized by N-capping interactions through aspartic acids or serines (green residues). The location of the helices within the molecular recognition element is shown in the primary sequence. Modified from ref. 100. (B) Ensemble description of MeV XD in complex with the 488–525 region of NTAIL. Model of the partly disordered MeV NTAIL–XD complex as a conformational ensemble representing the 50 best-fit structures of the 488–525 region of NTAIL in complex with XD. The NTAIL conformers are depicted with a color gradient ranging from yellow to red with increasing structural density, while XD is shown in black. Modified from ref. 119. | ||
The occurrence of a transiently populated α-helix even in the absence of the partner would suggest that the molecular mechanism governing the folding of NTAIL induced by XD could rely on conformer selection (i.e. selection by the partner of a pre-existing conformation).114,115 Recent data based on a quantitative analysis of NMR titration studies101 suggest however that the binding reaction may also imply a binding intermediate in the form of a weak, nonspecific encounter complex and hence may also occur through induced folding.116 That binding coupled to folding events rather may rely on a mixed mechanism implying both induced folding (i.e. folding after binding) and conformational selection (i.e. folding before binding) has already been reported.117
5. Residual flexibility within NiV, HeV and MeV NTAIL–XD complexes
SAXS studies provided a low-resolution model of the MeV NTAIL–XD complex, which showed that most of NTAIL (residues 401–488) remains disordered within the complex.28 As such, the MeV NTAIL–XD complex provides an illustrative example of “fuzziness”, where this term has been recently coined by Tompa and Fuxreiter to designate the persistence of conspicuous regions of disorder, often playing a functional role in binding, within protein complexes implicating IDPs.118 In further support of the residual flexibility in the bound form of MeV NTAIL, a novel approach that relies on a combination of SDSL EPR spectroscopy and modeling of local rotation conformational spaces showed that in spite of the local gain of rigidity induced by XD binding, the 505–525 region of NTAIL conserves a significant degree of freedom even in the bound form (Fig. 5B).119Similarly, HenipavirusNTAIL–XD complexes provide examples of “fuzziness” either. Indeed, the NMR behavior of HeV NTAIL–XD (i.e. vanishing of resonances during titration with no reappearance at saturation)23 suggests that even when bound to XD, NTAIL remains dynamic, undergoing exchange between different conformers at the XD surface.23 Moreover, the experimentally determined Stokes radius (RS) of the NiV NTAIL–XD complex (35.4 ± 3.1 Å) suggests that binding to XD does not imply formation of a compact complex (expected RS: 22.3 Å) and that it rather retains a considerable flexibility.23 In further support of the “fuzziness” within MeV, HeV and NiV NTAIL–XD complexes, the many observable and relatively sharp NMR resonances that are nearly unaltered upon addition of XD provide evidence that these NTAIL regions remain significantly disordered in the bound state.23,101
What is the functional role of such a fuzziness? We propose that the prevalently disordered nature of NTAIL even after complex formation may serve as a platform for the capture of other binding partners. In agreement, in the case of MeV NTAIL, Box1 has been shown to be responsible for the interaction with the cellular receptor NR,98,99,120 and Box3 was found to interact with the major inducible heat shock protein hsp70.121,122 In MeV, viral transcription and replication are enhanced by hsp70, with this stimulation relying on an interaction with NTAIL.121,123–129 Two binding sites for hsp70 have been identified within NTAIL:121,129 while the α-MoRE provides a high affinity-binding site (KD of 10 nM), a second low-affinity binding site is present within Box3.129,130 Since hsp70 was shown to competitively inhibit the binding of XD to NTAIL,121 it has been proposed that hsp70 could enhance transcription and genome replication by reducing the stability of P–NTAIL complexes, thereby promoting successive cycles of binding and release that are essential to polymerase movement along the nucleocapsid template.28,121 The hsp70-dependent reduction of the stability of P–NTAIL complexes would rely on competition between hsp70 and XD for binding to NTAIL through (i) competition for binding to the α-MORE (and this would occur at low hsp70 concentrations) and (ii) neutralization of the contribution of the C-terminus of NTAIL to the formation of a stable P–NTAIL complex (and this would occur in the context of elevated cellular levels of hsp70).121
In further support for a role of flexibility in the establishment of a broad molecular partnership and in agreement with previous reports emphasizing a relationship between disorder and protein interactivity,131–133MeV NTAIL was shown to interact with multiple partners. Indeed, beyond hsp70 and P, MeV NTAIL also interacts with the matrix protein134 and with other cellular proteins, including the interferon regulatory factor 3 (IRF3),135,136 the cell protein responsible for the nuclear export of N,137peroxiredoxin 1138 and possibly components of the cell cytoskeleton.139,140
6. Functional role of structural disorder within N and P in terms of transcription and replication
The elongation rate of the MeV polymerase was found to be rather slow (three nucleotides per s)141 in agreement with a rather stable NTAIL–XD complex (KD of 100 nM). Consistent with a model where a too much stable NTAIL–XD complex would hinder the polymerase processivity, the C-terminus of MeV NTAIL has been shown in minireplicon experiments to have an inhibitory role upon transcription and genome replication,129 probably reflecting its role in stabilizing the NTAIL–XD complex possibly through several weak, non-specific contacts with XD. According to this model, the interaction of Box3 with hsp 70 would stimulate transcription and replication by reducing the affinity of MeV NTAIL for XD, thereby promoting successive cycles of binding and release of the polymerase along the nucleocapsid template. A relatively labile complex can result either from a tight complex the strength of which is modulated by co-factors, as in the case of the MeV NTAIL–XD interaction, or from an inherently lower affinity of the binding reaction. SeV104 and Henipaviruses23 would provide examples of this latter scenario, with KD values in the μM range. Interestingly, in the case of RV, the KD between N-RNA rings and the C-terminal domain of the phosphoprotein was found to be 160 nM.142 In this case, a mechanism different from cartwheeling has been evoked, whereby the P protein would be permanently bound to the nucleocapsid template, and the polymerase would jump between adjacent P dimers.143The induced folding of NTAIL resulting from the interaction with P (and/or other physiological partners) could also exert an impact on the nucleocapsid conformation thereby favoring transcription and replication. This is indeed evidenced by electron microscopy analyses of MeV nucleocapsids formed by either N or NCORE indicating that the presence of MeV NTAIL was associated with a great degree of fragility (i.e.nucleocapsid flexibility).25,89,92,100 In the same vein, preliminary data indicate that incubation of MeV nucleocapsids in the presence of XD triggers an unwinding of the nucleocapsid, thus possibly enhancing the accessibility of genomic RNA to the polymerase complex (Bhella and Longhi, unpublished data). Similar studies, aimed at unveiling a possible similar phenomenon in Henipavirusnucleocapsids, are in progress.
The presence of unstructured domains on both N and P would allow for coordinated interactions between the polymerase complex and a large surface area of the nucleocapsid template, including successive turns of the helix. Indeed, the maximal extension of MeV PNT as measured by SAXS (Longhi and Receveur-Bréchot, unpublished data) is 40 nm. In comparison, one turn of the MeV nucleocapsid is 18 nm in diameter and 6 nm high.86 Thus PNT could easily stretch over several turns of the nucleocapsid, and since P is multimeric, N°–P might have a considerable extension. Likewise, the maximal extension of MeV NTAIL in solution is 13 nm.25 The very long reach of disordered regions could enable them to act as linkers and to tether partners on large macromolecular assemblies, thereby acting as scaffolding engines as already described for intrinsically disordered scaffold proteins.144,145
Finally, as binding of NTAIL to XD allows tethering of the L protein on the nucleocapsid template, the NTAIL–XD interaction is crucial for both viral transcription and genomic replication. Since neither NTAIL nor XD have cellular homologues, the NTAIL–XD interaction is an ideal target for antiviral inhibitors. Attempts at identifying small compounds able to block the HenipavirusNTAIL–XD interaction through screening of chemical libraries, are currently in progress.
7. Functional advantages of structural disorder within viral proteins
Recent bioinformatics studies showed that viruses and eukaryota have ten times more conserved disorder (roughly 1%) than archaea and bacteria (0.1%),146 and also pointed out that viral proteins, and in particular proteins from RNA viruses, are enriched in short disordered regions.147,148 In this latter studies, the authors propose that beyond affording a broad partnership, the wide occurrence of disordered regions in viral proteins could also be related to the typical high mutation rates of RNA viruses, representing a strategy for buffering the deleterious effects of mutations. Beyond these computational studies, a considerable body of experimental evidence has been collated pointing out the disordered nature of several viral proteins (or domains thereof).149Disorder has also been reported to provide a mean to tolerate insertions and/or deletions and to be thus abundant in region with dual coding capacity.150–154 Consistent with this relationship, when we compared the modular organization of the P proteins within the Paramyxovirinae subfamily,26 we noticed that a larger PNT domain in Henipaviruses accounts for the extra length of their P protein.21
In the same vein, PNT partially overlaps with the C protein (being encoded by the same RNA region) and the “spacer” region partially overlaps with the C-terminal domain of the V protein. The disordered nature of PNT and of the “spacer” region connecting PNT to PMD likely reflects a way of alleviating evolutionary constraints within overlapping reading frames. A similar scenario is found in the hepatitis C virus Core +1/S protein, which overlaps the Core protein gene in the +1 reading frame and has been reported to be intrinsically disordered.155 Disorder, which is encoded by a much wider portion of sequence space as compared to order, can indeed represent a strategy by which genes encoding overlapping reading frames can lessen evolutionary constraints imposed on their sequence by the overlap, allowing the encoded overlapping protein products to sample a wider sequence space without losing function.
Taking into account these considerations, as well as the correlation between overlapping genes and disorder, we propose that the main advantage of the abundance of disorder within viruses would reside in pleiotropy and genetic compaction. Indeed, disorder provides a solution to reduce both genome size and molecular crowding, where a single gene would (i) encode a single (regulatory) protein product that can establish multiple interactions via its disordered regions and hence exert multiple concomitant biological effects, and/or (ii) would encode more than one product by means of overlapping reading frames. In fact, since disordered regions are less sensitive to structural constraints than ordered ones, the occurrence of disorder within one or both protein products encoded by an overlapping reading frame can represent a strategy to alleviate evolutionary constraints imposed by the overlap. As such, disorder would confer to viruses the ability to “handle” overlaps, thus further expanding the coding potential of viral genomes.
Acknowledgements
We wish to thank all the persons who contributed to the works herein described. In particular, within the AFMB laboratory, we would like to thank Jean-Marie Bourhis, Benjamin Morin, Stéphane Gely, Stéphanie Costanzo, Stephanie Blangy, Laurent Mamelli, Matteo Colombo, Marie Couturier, Sabrina Rouger, Elodie Liquière, Bruno Canard, Kenth Johansson, David Karlin, François Ferron, Véronique Receveur-Brechot, Hervé Darbon, Cédric Bernard and Valérie Campanacci. We also thank our co-workers Michael Oglesbee (Ohio State University), Keith Dunker (Indiana University, USA), David Bhella (MRC, Glasgow, UK), Denis Gerlier (Lab. Virologie Humaine, Lyon, France), Hélène Valentin and Chantal Rabourdin-Combe (Lab. Immunobioogie Fondamentale et Clinique, Lyon, France), Gary Daughdrill (University of South Florida), Martin Blackledge and Malene Ringkjobing-Jensen (IBS, Grenoble, France), Rob W. H. Ruigrok and Marc Jamin (Unit of Virus Host Cell Interactions, Grenoble, France), André Fournel, Valérie Belle and Bruno Guigliarelli (BIP, CNRS, Marseille, France). We also want to thank Vladimir Uversky and Denis Gerlier for stimulating discussions. Finally, SL wishes to express her gratitude to Frédéric Carrière (EIPL, CNRS, Marseille, France) for having introduced her to EPR spectroscopy and for constant support. The studies mentioned in this review were carried out with the financial support of the European Commission, program RTD, QLK2-CT2001-01225, of the Agence Nationale de la Recherche, specific programs “Microbiologie et Immunologie” (ANR-05-MIIM-035-02) and “Physico-chimie du vivant” (ANR-08-PCVI-0020-01), and of the National Institute of Neurological Disorders and Stroke (R01 NS031693-11A2).References
- R. A. Lamb and G. D. Parks, Paramyxoviridae: the viruses and their replication, in Fields Virology, ed. D. M. Knipe and P. M. Howley, Lippincott Williams & Wilkins, Philadelphia, PA, 2007, 5th edn, pp. 1450–1497 Search PubMed.
- L. F. Wang, M. Yu, E. Hansson, L. I. Pritchard, B. Shiell, W. P. Michalski and B. T. Eaton, J. Virol., 2000, 74, 9972–9979 CrossRef CAS.
- B. T. Eaton, J. S. Mackenzie and L. F. Wang, Henipaviruses, in Fields Virology, ed. B. N. Fields, D. M. Knipe and P. M. Howley, Lippincott-Raven, Philadelphia, 2007, 5th edn, pp. 1587–1600 Search PubMed.
- M. Huber, R. Cattaneo, P. Spielhofer, C. Orvell, E. Norrby, M. Messerli, J. C. Perriard and M. A. Billeter, Virology, 1991, 185, 299–308 CrossRef CAS.
- D. Spehner, R. Drillien and P. M. Howley, Virology, 1997, 232, 260–268 CrossRef CAS.
- S. Longhi and B. Canard, Virologie, 1999, 3, 227–240 Search PubMed.
- R. A. Lamb and D. Kolakofsky, Paramyxoviridae: the viruses and their replication, in Fields Virology, ed. B. N. Fields, D. M. Knipe and P. M. Howley, Lippincott-Raven, Philadelphia, PA, 2001, 4th edn, pp. 1305–1340 Search PubMed.
- A. A. V. Albertini, G. Schoehn and R. W. Ruigrok, Virologie, 2005, 9, 83–92 Search PubMed.
- L. Roux, Virologie, 2005, 9, 19–34 Search PubMed.
- K. W. Ryan and A. Portner, Virology, 1990, 174, 515–521 CrossRef CAS.
- C. J. Buchholz, C. Retzler, H. E. Homann and W. J. Neubert, Virology, 1994, 204, 770–776 CrossRef CAS.
- M. Gopinath and M. S. Shaila, Virus Res., 2008, 135, 150–154 CrossRef CAS.
- T. Ogino, M. Kobayashi, M. Iwama and K. Mizumoto, J. Biol. Chem., 2005, 280, 4429–4435 CrossRef CAS.
- F. Ferron, S. Longhi, B. Henrissat and B. Canard, Trends Biochem. Sci., 2002, 27, 222–224 CrossRef CAS.
- A. A. Rahmeh, J. Li, P. J. Kranzusch and S. P. Whelan, J. Virol., 2009, 83, 11043–11050 CrossRef CAS.
- J. Li, A. Rahmeh, M. Morelli and S. P. Whelan, J. Virol., 2008, 82, 775–784 CrossRef CAS.
- J. M. Bourhis, B. Canard and S. Longhi, Virology, 2006, 344, 94–110 CrossRef CAS.
- J. M. Bourhis and S. Longhi, Measles virus nucleoprotein: structural organization and functional role of the intrinsically disordered C-terminal domain, in Measles virus nucleoprotein, ed. S. Longhi, Nova Publishers Inc., Hauppage, NY, 2007, pp. 1–35 Search PubMed.
- S. Longhi, Curr. Top. Microbiol. Immunol., 2009, 329, 103–128 CrossRef CAS.
- S. Longhi and M. Oglesbee, Protein Pept. Lett., 2010, 17, 961–978 CrossRef CAS.
- J. Habchi, L. Mamelli, H. Darbon and S. Longhi, PLoS One, 2010, 5, e11684 Search PubMed.
- Y. P. Chan, C. L. Koh, S. K. Lam and L. F. Wang, J. Gen. Virol., 2004, 85, 1675–1684 CrossRef CAS.
- J. Habchi, S. Blangy, L. Mamelli, M. Ringkjobing Jensen, M. Blackledge, H. Darbon, M. Oglesbee, Y. Shu and S. Longhi, J. Biol. Chem., 2011, 286, 13583–13602 CrossRef CAS.
- D. Karlin, S. Longhi, V. Receveur and B. Canard, Virology, 2002, 296, 251–262 CrossRef CAS.
- S. Longhi, V. Receveur-Brechot, D. Karlin, K. Johansson, H. Darbon, D. Bhella, R. Yeo, S. Finet and B. Canard, J. Biol. Chem., 2003, 278, 18638–18648 CrossRef CAS.
- D. Karlin, F. Ferron, B. Canard and S. Longhi, J. Gen. Virol., 2003, 84, 3239–3252 CrossRef CAS.
- J. Bourhis, K. Johansson, V. Receveur-Bréchot, C. J. Oldfield, A. K. Dunker, B. Canard and S. Longhi, Virus Res., 2004, 99, 157–167 CrossRef CAS.
- J. M. Bourhis, V. Receveur-Bréchot, M. Oglesbee, X. Zhang, M. Buccellato, H. Darbon, B. Canard, S. Finet and S. Longhi, Protein Sci., 2005, 14, 1975–1992 CrossRef CAS.
- J. M. Bourhis, B. Canard and S. Longhi, Virologie, 2005, 9, 367–383 Search PubMed.
- P. Radivojac, L. M. Iakoucheva, C. J. Oldfield, Z. Obradovic, V. N. Uversky and A. K. Dunker, Biophys. J., 2007, 92, 1439–1456 CrossRef CAS.
- V. N. Uversky and A. K. Dunker, Biochim. Biophys. Acta, 2010, 1804, 1231–1264 CAS.
- V. N. Uversky, Int. J. Biochem. Cell Biol., 2011, 43, 1090–1103 CrossRef CAS.
- P. Tompa, Structure and Function of Intrinsically Disordered Proteins, CRC Press, Taylor & Francis Group, Boca Raton, Florida, 2010.
- P. Tompa, Curr. Opin. Struct. Biol., 2011, 21, 419–425 CrossRef CAS.
- F. Ferron, S. Longhi, B. Canard and D. Karlin, Proteins, 2006, 65, 1–14 CrossRef CAS.
- J. M. Bourhis, B. Canard and S. Longhi, Curr. Protein Pept. Sci., 2007, 8, 135–149 CrossRef CAS.
- V. Receveur-Bréchot, J. M. Bourhis, V. N. Uversky, B. Canard and S. Longhi, Proteins: Struct., Funct., Bioinf., 2006, 62, 24–45 CrossRef.
- V. N. Uversky and S. Longhi, Instrumental analysis of intrinsically disordered proteins: assessing structure and conformation, John Wiley and Sons, Hoboken, New Jersey, 2010 Search PubMed.
- F. C. Gerard, A. Ribeiro Ede Jr., C. Leyrat, I. Ivanov, D. Blondel, S. Longhi, R. W. Ruigrok and M. Jamin, J. Mol. Biol., 2009, 388, 978–996 CrossRef CAS.
- M. T. Llorente, B. Barreno-Garcia, M. Calero, E. Camafeita, J. A. Lopez, S. Longhi, F. Ferron, P. F. Varela and J. A. Melero, J. Gen. Virol., 2006, 87, 159–169 CrossRef CAS.
- N. Tarbouriech, J. Curran, C. Ebel, R. W. Ruigrok and W. P. Burmeister, Virology, 2000, 266, 99–109 CrossRef CAS.
- N. Tarbouriech, J. Curran, R. W. Ruigrok and W. P. Burmeister, Nat. Struct. Biol., 2000, 7, 777–781 CrossRef CAS.
- V. N. Uversky, J. R. Gillespie and A. L. Fink, Proteins, 2000, 41, 415–427 CrossRef CAS.
- P. Lieutaud, B. Canard and S. Longhi, BMC Genomics, 2008, 9, S25 CrossRef.
- N. Watanabe, M. Kawano, M. Tsurudome, S. Kusagawa, M. Nishio, H. Komada, T. Shima and Y. Ito, J. Gen. Virol., 1996, 77, 327–338 CrossRef CAS.
- C. Leyrat, M. R. Jensen, E. A. Ribeiro Jr., F. C. Gerard, R. W. Ruigrok, M. Blackledge and M. Jamin, Protein Sci., 2011, 20, 542–556 CrossRef CAS.
- M. Chen, T. Ogino and A. K. Banerjee, J. Virol., 2007, 81, 13478–13485 CrossRef CAS.
- E. Garner, P. Romero, A. K. Dunker, C. Brown and Z. Obradovic, Genome Inform. Ser. Workshop Genome Inform., 1999, 10, 41–50 CAS.
- C. J. Oldfield, Y. Cheng, M. S. Cortese, P. Romero, V. N. Uversky and A. K. Dunker, Biochemistry, 2005, 44, 12454–12470 CrossRef CAS.
- A. Mohan, C. J. Oldfield, P. Radivojac, V. Vacic, M. S. Cortese, A. K. Dunker and V. N. Uversky, J. Mol. Biol., 2006, 362, 1043–1059 CrossRef CAS.
- V. Vacic, C. J. Oldfield, A. Mohan, P. Radivojac, M. S. Cortese, V. N. Uversky and A. K. Dunker, J. Proteome Res., 2007, 6, 2351–2366 CrossRef CAS.
- V. N. Uversky, Protein Sci., 2002, 11, 739–756 CrossRef CAS.
- A. K. Dunker, J. D. Lawson, C. J. Brown, R. M. Williams, P. Romero, J. S. Oh, C. J. Oldfield, A. M. Campen, C. M. Ratliff, K. W. Hipps, J. Ausio, M. S. Nissen, R. Reeves, C. Kang, C. R. Kissinger, R. W. Bailey, M. D. Griswold, W. Chiu, E. C. Garner and Z. Obradovic, J. Mol. Graphics Modell., 2001, 19, 26–59 CrossRef CAS.
- K. Dahlman-Wright and I. J. McEwan, Biochemistry, 1996, 35, 1323–1327 CrossRef CAS.
- Q. X. Hua, W. H. Jia, B. P. Bullock, J. F. Habener and M. A. Weiss, Biochemistry, 1998, 37, 5858–5866 CrossRef CAS.
- P. Tompa, Trends Biochem. Sci., 2002, 27, 527–533 CrossRef CAS.
- M. Fuxreiter, I. Simon, P. Friedrich and P. Tompa, J. Mol. Biol., 2004, 338, 1015–1026 CrossRef CAS.
- E. R. Lacy, I. Filippov, W. S. Lewis, S. Otieno, L. Xiao, S. Weiss, L. Hengst and R. W. Kriwacki, Nat. Struct. Mol. Biol., 2004, 11, 358–364 CAS.
- M. Chen, J. C. Cortay, I. R. Logan, V. Sapountzi, C. N. Robson and D. Gerlier, J. Virol., 2005, 79, 11824–11836 CrossRef CAS.
- A. Rahaman, N. Srinivasan, N. Shamala and M. S. Shaila, J. Biol. Chem., 2004, 279, 23606–23614 CrossRef CAS.
- I. Ivanov, T. Crepin, M. Jamin and R. W. Ruigrok, J. Virol., 2010, 84, 3707–3710 CrossRef CAS.
- H. Ding, T. J. Green, S. Lu and M. Luo, J. Virol., 2006, 80, 2808–2814 CrossRef CAS.
- K. Johansson, J. M. Bourhis, V. Campanacci, C. Cambillau, B. Canard and S. Longhi, J. Biol. Chem., 2003, 278, 44567–44573 CrossRef CAS.
- J. Habchi, L. Mamelli and S. Longhi, Structural disorder within the nucleoprotein and phosphoprotein from measles, Nipah and Hendra viruses, in Flexible viruses: structural disorder in viral proteins, ed. V. N. Uversky and S. Longhi, John Wiley and Sons, Hoboken, New Jersey, 2011, in press Search PubMed.
- R. L. Kingston, D. J. Hamel, L. S. Gay, F. W. Dahlquist and B. W. Matthews, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 8301–8306 CrossRef CAS.
- R. L. Kingston, L. S. Gay, W. S. Baase and B. W. Matthews, J. Mol. Biol., 2008, 379, 719–731 CrossRef CAS.
- L. Blanchard, N. Tarbouriech, M. Blackledge, P. Timmins, W. P. Burmeister, R. W. Ruigrok and D. Marion, Virology, 2004, 319, 201–211 CrossRef CAS.
- R. L. Kingston, A. B. Walter and L. S. Gay, J. Virol., 2004, 78, 8630–8640 CrossRef CAS.
- M. Mavrakis, A. A. McCarthy, S. Roche, D. Blondel and R. W. Ruigrok, J. Mol. Biol., 2004, 343, 819–831 CrossRef CAS.
- E. A. Ribeiro Jr., A. Favier, F. C. Gerard, C. Leyrat, B. Brutscher, D. Blondel, R. W. Ruigrok, M. Blackledge and M. Jamin, J. Mol. Biol., 2008, 382, 525–538 CrossRef.
- R. Assenberg, O. Delmas, J. Ren, P. O. Vidalain, A. Verma, F. Larrous, S. C. Graham, F. Tangy, J. M. Grimes and H. Bourhy, J. Virol., 2009, 84, 1089–1096 CrossRef.
- O. Delmas, R. Assenberg, J. M. Grimes and H. Bourhy, RNA Biol., 2010, 7, 322–327 CrossRef CAS.
- P. Bernado, L. Blanchard, P. Timmins, D. Marion, R. W. Ruigrok and M. Blackledge, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 17002–17007 CrossRef CAS.
- K. Houben, L. Blanchard, M. Blackledge and D. Marion, Biophys. J., 2007, 93, 2830–2844 CrossRef CAS.
- J. Curran, H. Homann, C. Buchholz, S. Rochat, W. Neubert and D. Kolakofsky, J. Virol., 1993, 67, 4358–4364 CAS.
- C. J. Buchholz, D. Spehner, R. Drillien, W. J. Neubert and H. E. Homann, J. Virol., 1993, 67, 5803–5812 CAS.
- B. Bankamp, S. M. Horikami, P. D. Thompson, M. Huber, M. Billeter and S. A. Moyer, Virology, 1996, 216, 272–277 CrossRef CAS.
- P. Liston, R. Batal, C. DiFlumeri and D. J. Briedis, Arch. Virol., 1997, 142, 305–321 CrossRef CAS.
- T. M. Myers, A. Pieters and S. A. Moyer, Virology, 1997, 229, 322–335 CrossRef CAS.
- T. M. Myers, S. Smallwood and S. A. Moyer, J. Gen. Virol., 1999, 80, 1383–1391 CAS.
- D. Karlin, S. Longhi and B. Canard, Virology, 2002, 302, 420–432 CrossRef CAS.
- D. Bhella, A. Ralph and R. P. Yeo, J. Mol. Biol., 2004, 340, 319–331 CrossRef CAS.
- M. G. Rudolph, I. Kraus, A. Dickmanns, M. Eickmann, W. Garten and R. Ficner, Structure (Camb), 2003, 11, 1219–1226 CrossRef CAS.
- D. Spehner, A. Kirn and R. Drillien, J. Virol., 1991, 65, 6296–6300 CAS.
- A. Warnes, A. R. Fooks, A. B. Dowsett, G. W. Wilkinson and J. R. Stephenson, Gene, 1995, 160, 173–178 CrossRef CAS.
- D. Bhella, A. Ralph, L. B. Murphy and R. P. Yeo, J. Gen. Virol., 2002, 83, 1831–1839 CAS.
- W. S. Tan, S. T. Ong, M. Eshaghi, S. S. Foo and K. Yusoff, J. Med. Virol., 2004, 73, 105–112 CrossRef CAS.
- Y. M. Kerdiles, B. Cherif, J. C. Marie, N. Tremillon, B. Blanquier, G. Libeau, A. Diallo, T. F. Wild, M. B. Villiers and B. Horvat, Viral Immunol., 2006, 19, 324–334 CrossRef CAS.
- G. Schoehn, M. Mavrakis, A. Albertini, R. Wade, A. Hoenger and R. W. Ruigrok, J. Mol. Biol., 2004, 339, 301–312 CrossRef CAS.
- M. H. Heggeness, A. Scheid and P. W. Choppin, Proc. Natl. Acad. Sci. U. S. A., 1980, 77, 2631–2635 CrossRef CAS.
- M. H. Heggeness, A. Scheid and P. W. Choppin, Virology, 1981, 114, 555–562 CrossRef CAS.
- A. Desfosses, G. Goret, L. Farias Estrozi, R. W. Ruigrok and I. Gutsche, J. Virol., 2011, 85, 1391–1395 CrossRef CAS.
- T. J. Green, X. Zhang, G. W. Wertz and M. Luo, Science, 2006, 313, 357–360 CrossRef CAS.
- A. A. Albertini, A. K. Wernimont, T. Muziol, R. B. Ravelli, C. R. Clapier, G. Schoehn, W. Weissenhorn and R. W. Ruigrok, Science, 2006, 313, 360–363 CrossRef CAS.
- M. Luo, T. J. Green, X. Zhang, J. Tsao and S. Qiu, Virus Res., 2007, 129, 246–251 CrossRef CAS.
- R. G. Tawar, S. Duquerroy, C. Vonrhein, P. F. Varela, L. Damier-Piolle, N. Castagné, K. MacLellan, H. Bedouelle, G. Bricogne, D. Bhella, J. F. Eleouet and F. A. Rey, Science, 2009, 326, 1279–1283 CrossRef CAS.
- A. Diallo, T. Barrett, M. Barbron, G. Meyer and P. C. Lefevre, J. Gen. Virol., 1994, 75(Pt 1), 233–237 CrossRef CAS.
- D. Laine, M. Trescol-Biémont, S. Longhi, G. Libeau, J. Marie, P. Vidalain, O. Azocar, A. Diallo, B. Canard, C. Rabourdin-Combe and H. Valentin, J. Virol., 2003, 77, 11332–11346 CrossRef CAS.
- D. Laine, J. Bourhis, S. Longhi, M. Flacher, L. Cassard, B. Canard, C. Sautès-Fridman, C. Rabourdin-Combe and H. Valentin, J. Gen. Virol., 2005, 86, 1771–1784 CrossRef CAS.
- M. Ringkjøbing Jensen, G. Communie, E. D. Ribeiro Jr., N. Martinez, A. Desfosses, L. Salmon, L. Mollica, F. Gabel, M. Jamin, S. Longhi, R. W. Ruigrok and M. Blackledge, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 9839–9844 CrossRef.
- S. Gely, D. F. Lowry, C. Bernard, M. Ringkjobing-Jensen, M. Blackledge, S. Costanzo, H. Darbon, G. W. Daughdrill and S. Longhi, J. Mol. Recognit., 2010, 23, 435–447 CrossRef CAS.
- C. Bernard, S. Gely, J. M. Bourhis, X. Morelli, S. Longhi and H. Darbon, FEBS Lett., 2009, 583, 1084–1089 CrossRef CAS.
- K. Yegambaram and R. L. Kingston, Protein Sci., 2010, 19, 893–899 CrossRef CAS.
- K. Houben, D. Marion, N. Tarbouriech, R. W. Ruigrok and L. Blanchard, J. Virol., 2007, 81, 6807–6816 CrossRef CAS.
- B. Meszaros, P. Tompa, I. Simon and Z. Dosztanyi, J. Mol. Biol., 2007, 372, 549–561 CrossRef CAS.
- T. Mittag, S. Orlicky, W. Y. Choy, X. Tang, H. Lin, F. Sicheri, L. E. Kay, M. Tyers and J. D. Forman-Kay, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 17772–17777 CrossRef CAS.
- S. C. Sue, C. Cervantes, E. A. Komives and H. J. Dyson, J. Mol. Biol., 2008, 380, 917–931 CrossRef CAS.
- R. Kiss, Z. Bozoky, D. Kovacs, G. Rona, P. Friedrich, P. Dvortsak, R. Weisemann, P. Tompa and A. Perczel, FEBS Lett., 2008, 582, 2149–2154 CrossRef CAS.
- B. Morin, J. M. Bourhis, V. Belle, M. Woudstra, F. Carrière, B. Guigliarelli, A. Fournel and S. Longhi, J. Phys. Chem. B, 2006, 110, 20596–20608 CrossRef CAS.
- V. Belle, S. Rouger, S. Costanzo, E. Liquiere, J. Strancar, B. Guigliarelli, A. Fournel and S. Longhi, Proteins: Struct., Funct., Bioinf., 2008, 73, 973–988 CrossRef CAS.
- V. Belle, S. Rouger, S. Costanzo, S. Longhi and A. Fournel, Site-directed spin labeling EPR spectroscopy, in Instrumental analysis of intrinsically disordered proteins: assessing structure and conformation, ed. V. N. Uversky and S. Longhi, John Wiley and Sons, Hoboken, New Jersey, 2010 Search PubMed.
- M. R. Jensen, K. Houben, E. Lescop, L. Blanchard, R. W. Ruigrok and M. Blackledge, J. Am. Chem. Soc., 2008, 130, 8055–8061 CrossRef CAS.
- L. Serrano and A. R. Fersht, Nature, 1989, 342, 296–299 CrossRef CAS.
- C. D. Tsai, B. Ma, S. Kumar, H. Wolfson and R. Nussinov, Crit. Rev. Biochem. Mol. Biol., 2001, 36, 399–433 CrossRef CAS.
- C. J. Tsai, B. Ma, Y. Y. Sham, S. Kumar and R. Nussinov, Proteins: Struct., Funct., Bioinf., 2001, 44, 418–427 CrossRef CAS.
- B. A. Shoemaker, J. J. Portman and P. G. Wolynes, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 8868–8873 CrossRef CAS.
- L. M. Espinoza-Fonseca, Biochem. Biophys. Res. Commun., 2009, 382, 479–482 CrossRef CAS.
- P. Tompa and M. Fuxreiter, Trends Biochem. Sci., 2008, 33, 2–8 CrossRef CAS.
- A. Kavalenka, I. Urbancic, V. Belle, S. Rouger, S. Costanzo, S. Kure, A. Fournel, S. Longhi, B. Guigliarelli and J. Strancar, Biophys. J., 2010, 98, 1055–1064 CrossRef CAS.
- D. Laine, P. Vidalain, A. Gahnnam, J. C. Cortay, D. Gerlier, C. Rabourdin-Combe and H. Valentin, Interaction of measles virus nucleoprotein with cell surface receptors: impact on cell biology and immune response, in Measles virus nucleoprotein, ed. S. Longhi, Nova Publishers Inc., Hauppage, NY, 2007, pp. 113–152 Search PubMed.
- X. Zhang, J. M. Bourhis, S. Longhi, T. Carsillo, M. Buccellato, B. Morin, B. Canard and M. Oglesbee, Virology, 2005, 337, 162–174 CrossRef CAS.
- M. Couturier, M. Buccellato, S. Costanzo, J. M. Bourhis, Y. Shu, M. Nicaise, M. Desmadril, C. Flaudrops, S. Longhi and M. Oglesbee, J. Mol. Recognit., 2010, 23, 301–315 CAS.
- T. Carsillo, X. Zhang, D. Vasconcelos, S. Niewiesk and M. Oglesbee, J. Virol., 2006, 80, 2904–2912 CrossRef CAS.
- M. Oglesbee, Nucleocapsid protein interactions with the major inducible heat shock protein, in Measles virus nucleoprotein, ed. S. Longhi, Nova Publishers Inc., Hauppage, NY, 2007, pp. 53–98 Search PubMed.
- M. J. Oglesbee, H. Kenney, T. Kenney and S. Krakowka, Virology, 1993, 192, 556–567 CrossRef CAS.
- M. J. Oglesbee, Z. Liu, H. Kenney and C. L. Brooks, J. Gen. Virol., 1996, 77, 2125–2135 CrossRef CAS.
- D. Vasconcelos, E. Norrby and M. Oglesbee, J. Gen. Virol., 1998, 79, 1769–1773 CAS.
- D. Y. Vasconcelos, X. H. Cai and M. J. Oglesbee, J. Gen. Virol., 1998, 79, 2239–2247 CAS.
- X. Zhang, C. Glendening, H. Linke, C. L. Parks, C. Brooks, S. A. Udem and M. Oglesbee, J. Virol., 2002, 76, 8737–8746 CrossRef CAS.
- T. Carsillo, Z. Traylor, C. Choi, S. Niewiesk and M. Oglesbee, J. Virol., 2006, 80, 11031–11039 CrossRef CAS.
- A. K. Dunker, M. S. Cortese, P. Romero, L. M. Iakoucheva and V. N. Uversky, FEBS J., 2005, 272, 5129–5148 CrossRef CAS.
- V. N. Uversky, C. J. Oldfield and A. K. Dunker, J. Mol. Recognit., 2005, 18, 343–384 CrossRef CAS.
- C. Haynes, C. J. Oldfield, F. Ji, N. Klitgord, M. E. Cusick, P. Radivojac, V. N. Uversky, M. Vidal and L. M. Iakoucheva, PLoS Comput. Biol., 2006, 2, e100 Search PubMed.
- M. Iwasaki, M. Takeda, Y. Shirogane, Y. Nakatsu, T. Nakamura and Y. Yanagi, J. Virol., 2009, 83, 10374–10383 CrossRef CAS.
- B. R. tenOever, M. J. Servant, N. Grandvaux, R. Lin and J. Hiscott, J. Virol., 2002, 76, 3659–3669 CrossRef CAS.
- M. Colombo, J. M. Bourhis, C. Chamontin, C. Soriano, S. Villet, S. Costanzo, M. Couturier, V. Belle, A. Fournel, H. Darbon, D. Gerlier and S. Longhi, Virol. J., 2009, 6, 59 CrossRef.
- H. Sato, M. Masuda, R. Miura, M. Yoneda and C. Kai, Virology, 2006, 352, 121–130 CrossRef CAS.
- A. Watanabe, M. Yoneda, F. Ikeda, A. Sugai, H. Sato and C. Kai, J. Virol., 2011, 85, 2247–2253 CrossRef.
- B. P. De and A. K. Banerjee, Microsc. Res. Tech., 1999, 47, 114–123 CrossRef CAS.
- S. A. Moyer, S. C. Baker and S. M. Horikami, J. Gen. Virol., 1990, 71, 775–783 CrossRef CAS.
- S. Plumet, W. P. Duprex and D. Gerlier, J. Virol., 2005, 79, 6900–6908 CrossRef CAS.
- E. D. Ribeiro Jr., C. Leyrat, F. C. Gerard, A. A. Albertini, C. Falk, R. W. Ruigrok and M. Jamin, J. Mol. Biol., 2009, 394, 558–575 CrossRef.
- C. Leyrat, F. C. Gerard, E. de Almeida Ribeiro Jr., I. Ivanov, R. W. Ruigrok and M. Jamin, Protein Pept. Lett., 2010, 17, 979–987 CrossRef CAS.
- M. S. Cortese, V. N. Uversky and A. K. Dunker, Prog. Biophys. Mol. Biol., 2008, 98, 85–106 CrossRef CAS.
- A. Balazs, V. Csizmok, L. Buday, M. Rakacs, R. Kiss, M. Bokor, R. Udupa, K. Tompa and P. Tompa, FEBS J., 2009, 276, 3744–3756 CrossRef CAS.
- J. W. Chen, P. Romero, V. N. Uversky and A. K. Dunker, J. Proteome Res., 2006, 5, 879–887 CrossRef CAS.
- N. Tokuriki, C. J. Oldfield, V. N. Uversky, I. N. Berezovsky and D. S. Tawfik, Trends Biochem. Sci., 2009, 34, 53–59 CrossRef CAS.
- B. Xue, R. W. Williams, C. J. Oldfield, G. K. Goh, A. K. Dunker and V. N. Uversky, Protein Pept. Lett., 2010, 17, 932–951 CrossRef CAS.
- V. N. Uversky and S. Longhi, Flexible viruses: structural disorder in viral proteins, John Wiley and Sons, Hoboken, New Jersey, 2011, in press Search PubMed.
- I. K. Jordan, B. A. Sutter and M. A. McClure, Mol. Biol. Evol., 2000, 17, 75–86 CAS.
- A. Narechania, M. Terai and R. D. Burk, J. Gen. Virol., 2005, 86, 1307–1313 CrossRef CAS.
- C. Rancurel, M. Khosravi, K. A. Dunker, P. R. Romero and D. Karlin, J. Virol., 2009, 83, 10719–10736 CrossRef CAS.
- P. R. Romero, S. Zaidi, Y. Y. Fang, V. N. Uversky, P. Radivojac, C. J. Oldfield, M. S. Cortese, M. Sickmeier, T. LeGall, Z. Obradovic and A. K. Dunker, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 8390–8395 CrossRef CAS.
- E. Kovacs, P. Tompa, K. Liliom and L. Kalmar, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 5429–5434 CrossRef CAS.
- A. Boumlic, Y. Nomine, S. Charbonnier, G. Dalagiorgou, N. Vassilaki, B. Kieffer, G. Trave, P. Mavromara and G. Orfanoudakis, FEBSJ., 2010, 277, 774–789 CrossRef CAS.
- M. A. Larkin, G. Blackshields, N. P. Brown, R. Chenna, P. A. McGettigan, H. McWilliam, F. Valentin, I. M. Wallace, A. Wilm, R. Lopez, J. D. Thompson, T. J. Gibson and D. G. Higgins, Bioinformatics, 2007, 23, 2947–2948 CrossRef CAS.
- P. Gouet, E. Courcelle, D. I. Stuart and F. Metoz, Bioinformatics, 1999, 15, 305–308 CrossRef CAS.
- K. Bryson, L. J. McGuffin, R. L. Marsden, J. J. Ward, J. S. Sodhi and D. T. Jones, Nucleic Acids Res., 2005, 33, W36–W38 CrossRef CAS.
- W. L. DeLano, Proteins: Struct., Funct., Bioinf., 2002, 30, 442–454 Search PubMed.
Footnote |
| † Published as part of a Molecular BioSystems themed issue on Intrinsically Disordered Proteins: Guest Editor M. Madan Babu. |
| This journal is © The Royal Society of Chemistry 2012 |