Role of an intrinsically disordered conformation in AMPK-mediated phosphorylation of ULK1 and regulation of autophagy†
Shagufta H.
Khan
and
Raj
Kumar
*
Department of Basic Sciences, The Commonwealth Medical College, 525 Pine Street, Rm 3034, Scranton, PA-18509, USA. E-mail: rkumar@tcmedc.org; Fax: +1 570-504-9660; Tel: +1 570-504-9675
First published on 18th August 2011
Abstract
Recently, it has been established that there is a direct link between adenosine monophosphate activated protein kinase (AMPK), which is an energy sensor and is activated by glucose starvation, and Unc-51-like kinase 1 (ULK1) in triggering autophagy. Proper phosphorylation of ULK1 is crucial for ULK1/AMPK association and subsequent ULK1 functions in response to nutrient deprivation. Signaling modulated viaphosphorylation often involves a flexible/unstructured or an intrinsically disordered (ID) region of proteins. Structural analyses of the ULK1 protein suggest that most of its functionally important phosphorylation sites are located in an ID region. We propose that this ID nature facilitates AMPK-mediated phosphorylation of ULK1, which may provide a mechanism for ULK1 functions in response to nutrient deprivation. Understanding how an ID region of ULK1 modulates its post-translational modifications through AMPK in regulating allosteric coupling will significantly help in defining the cellular and molecular mechanisms involved in ULK1/AMPK functions and in regulation of autophagy.
 Shagufta H. Khan | Shagufta Khan is currently working as a Senior Research Associate in the Department of Basic Sciences at The Commonwealth Medical College, Scranton, PA, USA. She received her Masters degree in Biotechnology from Aligarh University, Aligarh, India. Her area of research mainly involves the role of post-translational modifications-, protein–protein interactions-, and osmolyte-induced folding of an intrinsically disordered N-terminal activation function domain of the steroid hormone receptors in transcriptional regulation. To her credit, she has more than a dozen research publications in reputed scientific journals. |
 Raj Kumar | Dr Kumar is currently a Professor of Biochemistry at The Commonwealth Medical College (TCMC), Scranton, PA, USA. He received his PhD from University of Lucknow, Lucknow, India, and post doctoral training at University of Texas Medical Branch (UTMB), Galveston, TX, USA. Before moving to TCMC, he held the position of Associate Professor in the Department of Internal Medicine at UTMB. Dr Kumar's current area of research is focused on the role of intrinsically disordered activation domain of transcription factors in the gene regulation. Dr Kumar has more than 75 publications to his credit in reputed scientific journals, and a patent. |
Introduction
Autophagy is critical for maintaining cellular homeostasis, and the majority of long-lived proteins and organelles are degraded viaautophagy.1–5 Cellular stress including nutrient depletion, hypoxia, and/or oxidative damage markedly up-regulate autophagy.6–8 During the time of starvation, autophagy provides essential support for sustained cell survival. Autophagy is also required for removing protein aggregates and as an innate intracellular defense mechanism against various pathogens.9Autophagy is an evolutionarily conserved homeostatic process for the turnover of cellular contents, organelles and misfolded proteins through the lysosomal machinery.10–12 It has been shown that chaperone-mediated autophagy is implicated in the turnover of proteins that are prone to aggregation, suggesting a pivotal role of the autophagy in the pathophysiology of neurodegenerative diseases.13–15 Due to its pivotal role in survival, homeostasis, and development, autophagy has attracted intensive interest, and it has been implicated in a wide range of processes and diseases including cancer, immune response, development, and aging among others.15–18 In addition, autophagy-related disruption is implicated in the pathophysiology of a number of neurodegenerative diseases including Huntington's, Parkinson's, and Alzheimer's diseases in which protein misfolding and improper clearance of injurious protein aggregates play critical roles.19–21The molecular basis of autophagy involves the concerted activation of proteins encoded by the family of autophagy-related (Atg) genes.22–24 Among them, the serine/threonine protein kinase Atg1, which appears to be essential at the early stages of autophagy, is the best characterized one.25 In mammals, Unc-51-like kinase 1 and 2 (ULK1 and ULK2) are the most closely related members of the family, sharing high homology within their protein kinase domains.26,27 However, the specific function of ULK1 and ULK2 in mammalian autophagy is not fully understood. It has been known that protein phosphorylation events are central to the control of ULK1 activity.28Phosphorylation of ULK1 is mediated by mammalian target-of-rapamycin (mTOR) kinase and/or adenosine monophosphate activated protein kinase (AMPK).29,30AMPK interacts with ULK1 in a nutrient-dependent manner.30 Proper phosphorylation of ULK1 is crucial for ULK1/AMPK association.30ULK1 is known to form a stable complex with Atg13 and focal adhesion kinase (FAK) family interacting protein (FIP 200).31 This complex is negatively regulated by the mammalian target of mTORC1 in a nutrient-dependent way whereas AMPK stimulates autophagy by inhibiting mTORC1.30 Recently, it has been reported that AMPK interacts with ULK1, which results in site-specific phosphorylation of ULK1 and its activation, which in turn bypasses mTOR-inhibition that leads to AMPK–ULK1 mediated autophagy30 (Fig. 1). In this review article, we discuss the role of phosphorylation and intrinsically disordered (ID) conformation of ULK1 in the regulation of autophagy.
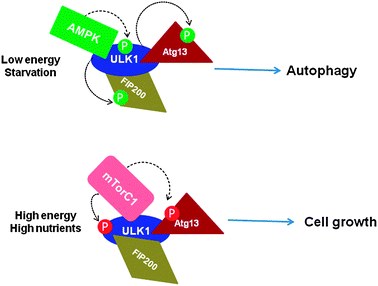 | ||
| Fig. 1 Phosphorylation-dependent ULK1–Atg13–FIP200 complex formation and regulation of autophagy (based on ref. 30 and 31). Under starvation/depleted energy conditions of cells, mTORC1 dissociates from the complex, ULK1 is phosphorylated by AMPK binding, which in turn phosphorylates Atg13 and FIP200, resulting in autophagy induction (upper panel). Under nutrient-rich conditions, mTORC1 binds to ULK1 and inhibits autophagy by phosphorylating ULK1 and Atg13, resulting in cell growth (lower panel). Arrows indicate the mediators of specific phosphorylation sites. P indicates phosphorylation. P in red circles indicates inhibitory sites whereas that in green indicates inducers. | ||
Role of ULK1 in the regulation of autophagy
Autophagy helps cells, organs, and entire organisms endure various stress conditions, such as limited nutrients, decreased energy supply, or low oxygen levels. Autophagy also plays crucial roles in development, innate immune defense, protein quality control, tumor suppression, and cell death.1–8 Under normal physiological conditions (when nutrients are abundant), basal levels of autophagy are mainly involved in degradation of long-lived proteins and turn-over of damaged cellular organelles.1–8 On the other hand, under stress conditions (such as nutrient starvation; low glucose levels), autophagy is induced to provide cells with additional internal nutrient supplies. This induction is largely due to inhibition of mTOR complex 1 and other nutrient-sensing signaling.32–34Under nutrient-rich conditions, Atg1 kinase induces autophagy upon starvation by forming an autophagy-initiating complex with Atg13, Atg17, Atg2, and Atg31, which trigger downstream events.30ULK1 interacts with several autophagy-related partners such as Atg13 and FIP200, which is a key regulatory step for autophagy initiation.35–41 The phosphorylation status of ULK1 plays a critical role in the formation of these complexes, and several phosphorylation sites in the ULK1 protein have been mapped.30AMPK, a key energy-surveillance kinase complex is known to directly associate with the ULK1 complex in a nutrient-dependent manner, and this association is dependent upon ULK1 phosphorylation.30 It has been proposed that the ULK1/AMPK interaction prepares cells to promptly initiate autophagy upon nutrient deprivation.30AMPK is a conserved metabolic switch that senses the cellular energy status and governs energy homeostasis through its regulation of glucose and lipid metabolism.42–44
Autophagy is generally initiated under nutrient-limited conditions by ULK1.45ULK1 acts at the initiation step of autophagy and forms a complex with mAtg13, FIP200, and Atg101.45 Recently, in the Jan 28, 2011, issue of Science, Egan et al.30 established a direct link between AMPK, which is an energy sensor and is activated by glucose starvation, and ULK1 in triggering autophagy. In this article authors reported that in mammals, phosphorylation of ULK1 by AMPK is required for ULK1 functions in response to nutrient deprivation.30 Using two-part screening, they identified four potential ULK1 phosphorylation sites (Ser467, Ser555, Thr574, and Ser637) in the central Ser/Pro Rich (SPR) domain.30Phosphorylation by AMPK often creates protein surfaces for binding partners to form stable complexes.45 In fact, authors reported several such binding partners for ULK1 that are dependent upon ULK1 phosphorylation by AMPK.30
AMPK-mediated phosphorylation of ULK1
AMPK is a positive regulator that stimulates autophagy in response to energy depletion, and recent studies suggest that AMPK directly regulates autophagy through phosphorylating and activating ULK1.28,30 On the other hand, the mTORC1 kinase complex (one of the primary negative regulators of autophagy) functions, in part, by phosphorylating and inhibiting ULK1 activity, and appears to prevent the physical interaction between AMPK and ULK1.28,30AMPK–ULK1 interaction is determined by the phosphorylation of ULK1 and consequent nutrient availability.30 This AMPK–ULK1 association may serve as a sequestering reservoir to confine ULK1 during normal growth conditions and prepare cells to promptly initiate autophagy under nutrient deprivation conditions.28,30The human ULK1 protein consists of 1051 amino acids and possesses a modular structure, which can be divided into three major functional domains (Fig. 2): an N-terminal kinase (KD), a central SPR, and a conserved C-terminal (CTD) domain. It is interesting to note that majority of phosphorylation sites including Ser467, Ser555, Thr574, and Ser637 that can be phosphorylated by AMPK are located within the SPR region.30 Co-immunoprecipitation analyses have revealed that AMPK binds to amino acids 654–828 within the SPR domain of ULK1.46 Together, it becomes quite evident that the SPR domain of ULK1 is the major target of AMPK-mediated phosphorylation and binding events. That raises the question: (i) whether SPR protein surfaces are well suited for its interaction with AMPK and subsequent phosphorylation of ULK1; (ii) if so, what is the conformation of the SPR domain; and (iii) whether ULK1–AMPK interaction/phosphorylation events influence the conformation of the SPR domain. It is important to note that compared to prokaryotes, eukaryotic genomes contain a much higher number of ID proteins, indicating toward greater need for signaling and regulation in nucleated cells.47
 | ||
| Fig. 2 A diagram of the human ULK1 protein showing major functional domains. KD = N-terminal kinase domain; SPR = central Ser/Pro rich domain; and CTD = conserved C-terminal domain. Numbers in black indicate the position of amino acid residues for each domain. Major functionally important phosphorylation sites in ULK1 due to AMPK (as reported by Egan et al., 201130) are also shown on the top. | ||
Intrinsically disordered domain/region and phosphorylation
It has been predicted that ID regions have a much higher frequency of known phosphorylation sites than ordered regions and that signalingviaphosphorylation-regulated protein–protein interactions often involves ID regions.48,49 In terms of structural consequences of site-specific phosphorylation, both disorder to order and order to disorder conformational transitions have been observed to follow the phosphorylation event.49 Conformational changes upon phosphorylation often affect protein functions. Site-specific phosphorylation is known to modulate the activity of numerous proteins involved in signal transduction, and regulates the binding affinity of transcription factors to their coactivators and DNA thereby altering gene expression, cell growth and differentiation.49 Since majority of the known phosphorylation sites in ULK1 are reported to be localized in the SPR domain, it will be interesting to find out the structural features of this domain and to compare with the structural features of other major domains of ULK1.However, the three-dimensional structure of the ULK1 protein is not known. Knowledge of the ULK1 structure/conformation will not only help us better understand the role of AMPK-mediated site-specific phosphorylation of ULK1 but it will also be a significant step towards understanding how AMPK/ULK1 interaction/phosphorylation regulates autophagy. It has been predicted that protein phosphorylation of Ser/Thr residues predominantly occurs within ID regions of signaling molecules rather than merely on surface residues.48,49 One of the main reasons for such propensity is to facilitate extensive formation of hydrogen binding between the backbones and/or side chains that can occur through disorder–order transition within the ID region.48,49 The formation of these hydrogen bonds would be difficult if the sites of phosphorylation were located within ordered regions.48,49 We have recently shown that site-specific phosphorylation of an ID region induces an ordered conformation in it such that protein surfaces are created for critical binding partners to form stable complexes.50
In this study, we found that an ID activation domain of the glucocorticoid receptor (an intracellular transcription factor) adopts a functionally folded conformation due to its site-specific phosphorylation by p38 mitogen-activated protein kinase, which is involved in apoptotic and gene-inductive events initiated by the glucocorticoid/glucocorticoid receptor. Further, this site-specific phosphorylation-induced secondary and tertiary structure formation specifically creates protein surfaces for interaction with critical coregulatory proteins and subsequently glucocorticoid receptor's transcriptional activity. These data demonstrate a mechanism through which ID activation domains of the steroid receptors and other similar transcription factors may adopt a functionally active conformation under physiological conditions. Based on these findings, we propose that under physiological conditions, site-specific phosphorylation plays a crucial role in allowing the ID domains to adopt functionally active conformation(s) in vivo. The resulting structurally modified forms of an ID region suit it for its varied interactions with other critical coregulatory proteins essential for function (Fig. 3). Since several protein kinase pathways are involved in cell signaling leading to diverse cellular responses, phosphorylation-induced conformational changes observed in the present study should provide a potential mechanism through which ID domains of signaling molecules exert their biological effects.
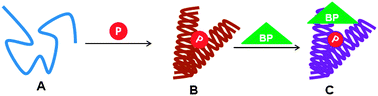 | ||
| Fig. 3 A general working model of an ID domain/region (A) of a protein undergoing conformational changes due to site-specific phosphorylation (B) that leads to its interaction with one or more binding partners (BPs) adapting a functionally active ordered conformation (C). | ||
Characterization of phosphorylation-induced structure formation in facilitating protein–protein interactions must be of great importance in understanding the mechanism by which AMPK regulates the functions of ULK1. We hypothesize that the SPR domain of ULK1 possesses a flexible structure such that it can adopt various configurations under different physiological conditions. This conformational flexibility, in turn, may allow the creation of protein surfaces, which can include or exclude various ULK1 binding partners. We further hypothesize that site-specific phosphorylation of residues within the SPR domain results in conformational changes to create specific interacting surfaces. This phenomenon is supported by the fact that under specific conditions, ULK1 either includes or excludes AMPK and mTOR complexes, which further dictates the interactions of other coregulatory proteins, essential for ULK1 functions. However, as yet, there are no structural data available to support this notion. Therefore, it is important to analyze the structural features of ULK1 particularly within the SPR domain.
Ser/Pro Rich (SPR) domain of ULK1 is intrinsically disordered
Since it has been hypothesized that protein phosphorylation of the Ser/Thr residue predominantly occurs within ID regions of signaling molecules rather than merely on surface residues, we determined whether the SPR domain of ULK1, which possesses most of the functionally known phosphorylation sites, exists in an ID conformation. To determine structural features of the ULK1 protein, we performed secondary structural analysis of ULK1 using Network Protein Sequence Analysis (http://npsa-pbil.ibcp.fr) as described in ref. 51. Secondary structural analysis of the ULK1 protein reveals that overall approximately half of the amino acid sequences show a globular structure with a significant amount (33%) of helical contents (Fig. 4a). When individual domains were analyzed, we observed that CTD consists of 87% helix, suggesting that this domain adopts a compact helical structure (Fig. 4d). Similar analysis for the KD domain revealed that this domain also possesses a significant amount of helical content (41%) suggesting that the KD domain is also relatively structured (Fig. 4b). On the other hand, the SPR domain showed mostly random coil (87%) configuration (Fig. 4c). The results are summarized in Table 1. Taken together, these analyses suggest that both KD and CTD regions possess significantly well-defined secondary structural elements whereas the SPR region is mostly unstructured.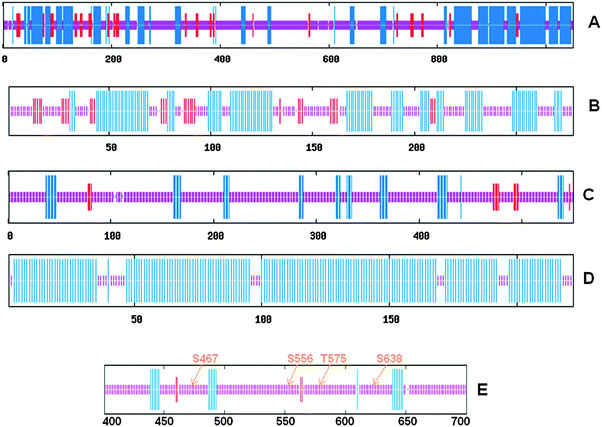 | ||
| Fig. 4 Consensus protein secondary structural elements predictions determined using Network Protein Sequence Analysis (http://npsa-pbil.ibcp.fr) as described in ref. 51 of the full length ULK1 (A), KD (B), SPR (C), CTD (D), and the region (amino acids 400–700) where most of the known phosphorylation sites are located (E). Blue, red, and purple colors indicate helix, β-sheet, and random coil, respectively. Numbers at the bottom of each panel indicate the position of amino acid residues of the ULK1 sequence. | ||
| Full-length | KD | SPR | CTD | a.a. 400–700 | |
|---|---|---|---|---|---|
| α-Helix | 33.2 | 41.4 | 11.8 | 87.3 | 8.3 |
| β-Sheet | 6.6 | 11.5 | 2.9 | 0.0 | 1.3 |
| Random coil | 56.8 | 44.6 | 84.4 | 12.6 | 87.7 |
Lack of a well-defined structure is generally associated with specific sites of enzyme-catalyzed phosphorylation and ubiquitination sites,52 which facilitates the inclusion or exclusion of specific binding partners in the complex.52,53 Because a significant part of the binding energy is required to fold an ID region, high specificity coupled with low affinity provides the basis for easy reversible interactions.48,54 Thus, flexibility and lack of structure in the SPR region may be a key feature for the ULK1 signaling conduit such that an ID state is needed before its association with AMPK. Since major AMPK-mediated phosphorylation sites (Ser467, Ser555, Thr574, and Ser637) of ULK1 are located within amino acid residues 400–700 (Fig. 4e), we determined whether this region possesses characteristics of an ID conformation, often found in signaling proteins with high propensity of functionally known phosphorylation sites. Predictions for the degree of disordered profiles were performed using Predictor of Naturally Disordered Regions (PONDR) prediction (www.pondr.com), as described in ref. 55 and 56. PONDR analysis for ID prediction confirms the ID nature of this region (Fig. 5a) as evident from the PONDR score obtained (more than 0.5). Similar results were obtained using Prediction of Intrinsically Unstructured Proteins (IUPred) analysis (Fig. 5b). IUPred analysis was performed (http://iupred.enzim.hu) as described in ref. 57. Together, these results suggest that the SPR region of ULK1, which possesses most of known phosphorylation sites, exists in an ID conformation.
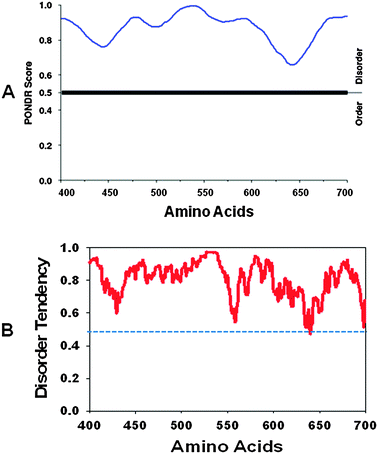 | ||
| Fig. 5 (A) PONDR plot for amino acids 400–700 of ULK1 protein disorder prediction (www.pondr.com) as described in ref. 55 and 56. The X-axis shows amino acid numbers in the ULK1 protein sequences, and the Y-axis shows the probability score. (B) IUPred plot for ULK1 protein (amino acids 400–700) disorder prediction (http://iupred.enzim.hu) as described in ref. 57. Scores above 0.5 are considered to be disordered sequences. | ||
It is a well accepted fact that generally, though perhaps not universally, under physiological conditions proteins must have a specific structure to carry out their proper functions. In the context of ULK1, it could be hypothesized that the SPR domain may be structured in vivo, at least when phosphorylated and/or directly involved in interactions with its binding partner proteins. Conformational uniqueness of most proteins determines their biological functions, and we propose that the ID nature of ULK1's SPR domain can explain at least, in part, the mechanism through which AMPK-mediated phosphorylation regulates autophagy under specific cellular conditions. The ID SPR domain can be thought of as a large collection of rapidly inter-convertible conformers, which can be selected from among available binding partner proteins to form the basis for building large complexes. Such complexes can dissociate and re-associate with differing compositions under a specific cellular environment.
Summary and perspectives
In recent years, it has become quite evident that numerous protein regions/domains exist as an ID protein. AMPK-mediated phosphorylation in turn leads to structure formation in the otherwise ID SPR region, which could result in structural rearrangements in ULK1 through intra-molecular cross-communications such that ULK1 protein surfaces are available for critical binding partner proteins including mAtg13, FIP200, and Atg101 (Fig. 6). On the other hand, ID nature could promote dissociation of mTOR under specific conditions such as starvation. This can be explained in terms of molecular crowding effects in cells on the dynamic structure of the ULK1 protein. It is important to note that under nutrient deficient conditions certain critical cellular components are compromised, affecting molecular crowding effects on protein structures. Under such circumstances, AMPK-mediated phosphorylation can induce a structure in ULK1 such that it can attract critical binding partners required for function. The elucidation of the dynamic interplay among AMPK, mTORC1, and the ULK1 complex and possibly other signaling pathways involving sensors will provide further information about the interconnected network of autophagy regulation. In this context, understanding how an ID region of ULK1 modulates its post-translational modifications through AMPK in regulating allosteric coupling will significantly help in defining the cellular and molecular mechanisms involved in ULK1/AMPK functions in response to nutrient deprivation.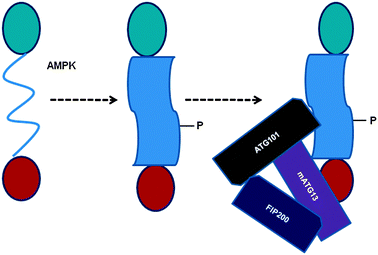 | ||
| Fig. 6 A mechanistic model for the AMPK-mediated phosphorylation-induced conformational changes in ULK1, which facilitates ULK1's interactions with specific protein binding partners. P indicates the phosphorylated state and the shape of the SPR domain represents conformational changes (from ID to globular form) due to AMPK-mediated phosphorylation. | ||
References
- N. Chen and V. Karantza, Cancer Biol. Ther., 2011, 11, 157–168 CrossRef CAS.
- A. Fleming, T. Noda, T. Yoshimori and D. C. Rubinsztein, Nat. Chem. Biol., 2011, 7, 9–17 CrossRef CAS.
- J. D. Rabinowitz and E. White, Science, 2010, 330, 1344–1348 CrossRef CAS.
- N. Mizushima and B. Levine, Nat. Cell Biol., 2010, 12, 823–830 CrossRef CAS.
- Z. Yang and D. J. Klionsky, Nat. Cell Biol., 2010, 12, 814–822 CrossRef CAS.
- S. Carloni, G. Buonocore and W. Balduini, Neurobiol. Dis., 2008, 32, 329–339 CrossRef CAS.
- J. M. Barth, J. Szabad, E. Hafen and K. Köhler, Cell Death Differ., 2011, 18, 915–924 CrossRef CAS.
- E. E. Essick and F. Sam, Oxidative Medicine and Cellular Longevity, 2010, 3, 168–177 CrossRef.
- B. Levine and V. Deretic, Nat. Rev. Immunol., 2007, 7, 767–777 CrossRef CAS.
- F. M. Menzies, K. Moreau and D. C. Rubinsztein, Curr. Opin. Cell Biol., 2011, 23, 190–197 CrossRef CAS.
- A. Wyttenbach, S. Hands, M. A. King, K. Lipkow and A. M. Tolkovsky, Autophagy, 2008, 4, 542–545 CAS.
- B. Ravikumar, S. Sarkar, J. E. Davies, M. Futter, M. Garcia-Arencibia, Z. W. Green-Thompson, M. Jimenez-Sanchez, V. I. Korolchuk, M. Lichtenberg, S. Luo, D. C. Massey, F. M. Menzies, K. Moreau, U. Narayanan, M. Renna, F. H. Siddiqi, B. R. Underwood, A. R. Winslow and D. C. Rubinsztein, Physiol. Rev., 2010, 90, 1383–1435 CrossRef CAS.
- S. Kaushik, U. Bandyopadhyay, S. Sridhar, R. Kiffin, M. Martinez-Vicente, M. Kon, S. J. Orenstein, E. Wong and A. M. Cuervo, J. Cell Sci., 2011, 124, 495–499 CrossRef CAS.
- B. Li, Y. Zhang, Y. Yuan and N. Chen, Parkinsonism Relat. Disord., 2011, 17, 231–235 CrossRef.
- H. Koga and A. M. Cuervo, Neurobiol. Dis., 2011, 43, 29–37 CrossRef CAS.
- F. Janku, D. J. McConkey, D. S. Hong and R. Kurzrock, Nat. Rev. Clin. Oncol., 2011, 16, 327–335 Search PubMed.
- T. Weide and T. B. Huber, Cell Tissue Res., 2011, 343, 467–473 CrossRef.
- T. Saitoh and S. Akira, J. Cell Biol., 2010, 189, 925–935 CrossRef CAS.
- P. I. Moreira, R. X. Santos, X. Zhu, H. G. Lee, M. A. Smith, G. Casadesus and G. Perry, Expert Rev. Neurother., 2010, 10, 1209–1218 CrossRef.
- M. Martinez-Vicente, Z. Talloczy, E. Wong, G. Tang, H. Koga, S. Kaushik, R. de Vries, E. Arias, S. Harris, D. Sulzer and A. M. Cuervo, Nat. Neurosci., 2010, 13, 567–576 CrossRef CAS.
- V. Choubey, D. Safiulina, A. Vaarmann, M. Cagalinec, P. Wareski, M. Kuum, A. Zharkovsky and A. Kaasik, J. Biol. Chem., 2011, 286, 10814–10824 CrossRef CAS.
- S. Meschini, M. Condello, P. Lista and G. Arancia, Curr. Cancer Drug Targets, 2011, 11, 357–379 CrossRef CAS.
- J. Geng and D. J. Klionsky, EMBO Rep., 2008, 9, 859–864 CrossRef CAS.
- Z. Xie and D. J. Klionsky, Nat. Cell Biol., 2007, 9, 1102–1109 CrossRef CAS.
- H. Nakatogawa, K. Suzuki, Y. Kamada and Y. Ohsumi, Nat. Rev. Mol. Cell Biol., 2009, 10, 458–467 CrossRef CAS.
- J. Yan, H. Kuroyanagi, T. Tomemori, N. Okazaki, K. Asato, Y. Matsuda, Y. Suzuki, Y. Ohshima, S. Mitani, Y. Masuho, T. Shirasawa and M. Muramatsu, Oncogene, 1999, 18, 5850–5859 CrossRef CAS.
- X. Zhou, J. R. Babu, S. da Silva, Q. Shu, I. A. Graef, T. Oliver, T. Tomoda, T. Tani, M. W. Wooten and F. Wang, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 5842–5847 CrossRef CAS.
- J. Kim, M. Kundu, B. Viollet and K. L. Guan, Nat. Cell Biol., 2011, 13, 132–141 CrossRef CAS.
- E. Y. Chan, Sci. Signaling, 2009, 2, pe51 CrossRef.
- D. F. Egan, D. B. Shackelford, M. M. Mihaylova, S. Gelino, R. A. Kohnz, W. Mair, D. S. Vasquez, A. Joshi, D. M. Gwinn, R. Taylor, J. M. Asara, J. Fitzpatrick, A. Dillin, B. Viollet, M. Kundu, M. Hansen and R. J. Shaw, Science, 2011, 331, 456–461 CrossRef CAS.
- D. G. Hardie, EMBO J., 2011, 30, 634–635 CrossRef CAS.
- C. He and D. J. Klionsky, Annu. Rev. Genet., 2009, 43, 67–93 CrossRef CAS.
- Y. Kamada, T. Funakoshi, T. Shintani, K. Nagano, M. Ohsumi and Y. Ohsumi, J. Cell Biol., 2000, 150, 1507–1513 CrossRef CAS.
- Y. Kamada, K. Yoshino, C. Kondo, T. Kawamata, N. Oshiro, K. Yonezawa and Y. Ohsumi, Mol. Cell. Biol., 2010, 30, 1049–1058 CrossRef CAS.
- E. Y. Chan, A. Longatti, N. C. McKnight and S. A. Tooze, Mol. Cell. Biol., 2009, 29, 157–171 CrossRef CAS.
- Y. Y. Chang and T. P. Neufeld, Mol. Biol. Cell, 2009, 20, 2004–2014 CrossRef CAS.
- I. G. Ganley, H. Lam du, J. Wang, X. Ding, S. Chen and X. Jiang, J. Biol. Chem., 2009, 284, 12297–12305 CrossRef CAS.
- N. Hosokawa, T. Hara, T. Kaizuka, C. Kishi, A. Takamura, Y. Miura, S. Iemura, T. Natsume, K. Takehana, N. Yamada, J. L. Guan, N. Oshiro and N. Mizushima, Mol. Biol. Cell, 2009, 20, 1981–1991 CrossRef CAS.
- N. Hosokawa, T. Sasaki, S. Iemura, T. Natsume, T. Hara and N. Mizushima, Autophagy, 2009, 5, 973–979 CrossRef CAS.
- C. H. Jung, C. B. Jun, S. H. Ro, Y. M. Kim, N. M. Otto, J. Cao, M. Kundu and D. H. Kim, Mol. Biol. Cell, 2009, 20, 1992–2003 CrossRef CAS.
- C. A. Mercer, A. Kaliappan and P. B. Dennis, Autophagy, 2009, 5, 649–662 CrossRef CAS.
- Y. C. Long and J. R. Zierath, J. Clin. Invest., 2006, 116, 1776–1783 CrossRef CAS.
- B. Viollet and F. Andreelli, Handb. Exp. Pharmacol., 2011, 203, 303–330 CrossRef CAS.
- D. B. Shackelford and R. J. Shaw, Nat. Rev. Cancer, 2009, 9, 563–575 CrossRef CAS.
- N. Mizushima, Curr. Opin. Cell Biol., 2010, 22, 132–139 CrossRef CAS.
- J. W. Lee, S. Park, Y. Takahashi and H. G. Wang, PLoS One, 2010, 5, e15394 Search PubMed.
- K. Sugase, H. J. Dyson and P. E. Wright, Nature, 2007, 447, 1021–1027 CrossRef CAS.
- A. K. Dunker and V. N. Uversky, Nat. Chem. Biol., 2008, 4, 229–230 CrossRef CAS.
- L. M. Iakoucheva, P. Radivojac, C. J. Brown, T. R. O'Connor, J. G. Sikes, Z. Obradovic and A. K. Dunker, Nucleic Acids Res., 2004, 32, 1037–1049 CrossRef CAS.
- A. S. Garza, S. H. Khan and R. Kumar, Mol. Cell. Biol., 2010, 30, 220–230 CrossRef CAS.
- C. Combet, C. Blanchet and C. Geourjon, Trends Biochem. Sci., 2000, 25, 147–150 CrossRef CAS.
- T. Zor, B. M. Mayr, H. J. Dyson, M. R. Montminy and P. E. Wright, J. Biol. Chem., 2002, 277, 42241–42248 CrossRef CAS.
- M. Grimmler, Y. Wang, T. Mund, Z. Cilensek, E. M. Keidel, M. B. Waddell, H. Jäkel, M. Kullmann, R. W. Kriwacki and L. Hengst, Cell (Cambridge, Mass.), 2007, 128, 269–280 CAS.
- A. A. Russo, P. D. Jeffrey, A. K. Patten, J. Massague and N. P. Pavletich, Nature, 1996, 382, 325–331 CrossRef CAS.
- P. Romero, Z. Obradovic, X. Li, E. C. Garner, C. J. Brown and A. K. Dunker, Proteins: Struct., Funct., Genet., 2001, 42, 38–48 CrossRef CAS.
- V. N. Uversky, J. R. Gillespie and A. L. Fink, Proteins: Struct., Funct., Genet., 2000, 41, 415–427 CrossRef CAS.
- Z. Dosztányi, V. Csizmók, P. Tompa and I. Simon, Bioinformatics, 2005, 21, 3433–3434 CrossRef.
Footnote |
| † Published as a part of a Molecular BioSystems themed issue on Intrinsically Disordered Proteins. Guest editor M. Madan Babu. |
| This journal is © The Royal Society of Chemistry 2012 |