DOI:
10.1039/C1MB05239J
(Paper)
Mol. BioSyst., 2012,
8, 237-246
Aromatic residues link binding and function of intrinsically disordered proteins†‡
Received
15th June 2011
, Accepted 29th July 2011
First published on 24th August 2011
Abstract
We have searched for intermolecular aromatic pairs in 77 protein–protein complexes of intrinsically disordered proteins (IDPs) to understand the role of π–π interactions in protein–protein interactions involving IDPs. We found that 40% of the complexes possess at least one intermolecular pair of aromatic residues. Analysis of composition, characteristics, location and the contribution to the free energy of binding showed that π–π interactions substantially contribute to binding by working as anchor residues, conformational locks, and ready-made recognition motifs required for specific binding. By using available experimental data we show that π–π interactions play a variety of roles that link binding of IDPs and their function in the cell. The results presented in this study pave the way to understand in atomic detail the inner workings of IDPs interaction networks.
Introduction
Protein–protein interactions (PPIs) are of utmost importance in the cellular machinery, as they are involved in a large number of functions such as signaling, transcription, and replication.1 Given the availability of high-resolution structures of protein–protein complexes, it has been possible to systematically study the connection between the nature of PPIs with their function in atomic detail.2–7 These studies have shown the importance of several factors for the formation and stability of PPIs, such as amino acid composition, geometric complementarity, solvent accessibility, secondary structure, electrostatic interactions, hydrophobic contacts, hydrogen bonding networks, π–π and π–cation interactions, among others.8–10 Among these factors, π–π interactions (i.e., interactions between aromatic residues)11–13 are particularly interesting not only because they occur frequently in protein–protein interfaces, but also because they play an important role in the recognition and specificity of PPIs.10,14
Although the importance of π–π interactions is well documented for PPIs between globular proteins, there is very limited information on their role in PPIs involving intrinsically disordered proteins (IDPs). Unlike globular proteins, IDPs do not possess a well-defined structure, and therefore they sample a very large number of different conformations in solution;15–17 however, this lack of structure is a feature that provides IDPs with several functional advantages over globular proteins.18–25 The inherent flexibility of IDPs has posed a challenge to researchers who have tried to understand the precise nature of conformational ensembles of IDPs in solution.26–30 Nevertheless, because many IDPs become partially ordered upon binding, it has been possible to obtain several atomic-resolution structures of their complexes with other proteins. Based on these structures and the inherent properties of IDPs, it has been found that these proteins use short sequential recognition elements for binding, usually known as primary contact sites, preformed structural elements or molecular recognition elements/features (MorEs/MorFs).31–34 Unlike globular proteins, IDPs also use a single continuous segment for binding;35 furthermore, it has been suggested that, in general, IDPs rely on hydrophobic–hydrophobic than on polar–polar interactions.35 Together, these generalizations have been used in predicting and understanding binding regions of IDPs.36,37 Despite these efforts, the contribution of π–π interactions in connecting binding and function of IDPs remains poorly understood. Interactions between aromatic residues are particularly interesting when studying PPIs of IDPs because of several reasons: (a) a vast majority of MorEs/MorFs contain at least one aromatic residue, phenylalanine being the most commonly occurring one;34 (b) although aromatic residues phenylalanine, tyrosine and tryptophan occur at lower frequencies in IDPs compared to ordered proteins, they appear to be highly conserved. Based on these observations, it was suggested that aromatic residues are important for function;38 (c) π–π interactions can provide both specificity and stability to protein–protein complexes;39 (d) π–π interactions are fundamentally different compared to purely hydrophobic interactions; for instance, Phe–Phe interactions in a β-hairpin were found to be enthalpically favorable and entropically unfavorable;40 and (e) the outputs of disorder predictors have been used to identify possible binding sites for partner proteins within disordered regions. In head-to-head comparisons, PONDR VL-XT outperformed RONN, IUPRED, PONDR VL3, DisPro, DRIPRED, and DISOPRED for this application.41 What is interesting here is dips from disorder towards order in the PONDR VL-XT plot is the indicator of a likely binding site, and, given the heavy use of aromatics as indicators of the structure, such dips usually arise when a region of disorder contains aromatic residues.42 These observations suggest that π–π interactions could play an important functional role in IDPs. Therefore, in this study we searched for intermolecular π–π interactions between IDPs and their binding partners in 77 protein–protein complexes previously identified by Mészáros et al.35,37 We further analyzed the composition, characteristics, location and contribution of aromatic residues to the free energy of binding of π–π interactions. Finally, by using experimental data we show that π–π interactions play a variety of roles that link binding and function of several IDPs.
Methods
Database selection
The dataset of protein–protein complexes involving IDPs consisted of 77 structures previously identified by Dosztányi and co-workers (Table S1, ESI‡).35,37 We used this dataset in this study as it was carefully constructed using experimental verification of both the disordered status in isolation and the formation of an ordered structure upon binding.37 The dataset also contained IDPs with both short disorder (chain length of 10–30 residues) and long disorder (chain length > 30 residues).37 The three-dimensional structures of the complexes were retrieved from the Protein Data Bank.43 Prior to analysis of the complexes, the structures determined using X-ray crystallography were subjected to energy minimization using the program NAMD.44
Identification of interactions involving aromatic residues
We searched for intermolecular interactions involving the side chain of aromatic residues (phenylalanine, tyrosine, tryptophan and histidine) using the structural definitions described below. T-shaped π–π interactions are geometrically defined by a centroid–centroid distance (Rcen) of ≤7.5 Å and a normal–normal angle (θ) that satisfies 86° ≤ θ ≤ 90°. On the other hand, stacking π–π interactions are defined as those satisfying a Rcen ≤ 7.5 and 0° ≤ θ ≤ 85°. As π–π stacking interactions can exist in a variety of geometrical arrangements (parallel and herringbone, Scheme 1), other parameters such as closest contact distances (Rclo) and normal-center angle (γ) were considered to accurately differentiate T-shaped and stacking geometries (Scheme 1).13 In addition, we included the non-classical hydrogen bond (X–H⋯π) in our search engine. In this case, X–H⋯π is considered to be present if the donor (i.e., N–H of tryptophan/histidine or O–H of tyrosine) and the center of the aromatic ring are separated by a distance of ≤4.5 Å and a hydrogen bond angle of ≤25°. We searched for the interactions satisfying these definitions using the program VMD.45
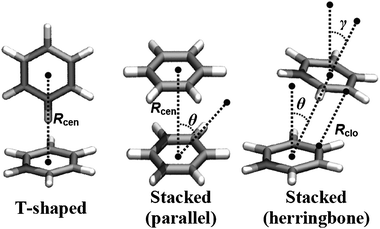 |
| | Scheme 1 Geometric definition of the π–π interactions considered in this study. For clarity, we show the benzene dimer as a model for π–π interactions. The definition of Rcen, Rclo, θ and γ is described in the Methods section. | |
Computational alanine scanning
To determine to what extent aromatic residues contribute to the free energy of binding, computational alanine scanning was performed using a program developed by Kortemme and Baker.46,47 This program identifies all residues in a protein–protein interface and computes the changes in binding free energy for alanine mutations (ΔΔGbind). Despite the simplicity and inherent limitations of the method, a recent study showed that it was able to qualitatively and quantitatively identify the residues that contribute to the free energy of binding of an IDP.48 The method uses a free energy function consisting of Lennard-Jones and hydrogen bond potentials as well as Coulomb electrostatics and an implicit solvation model. A detailed description of the method can be found in ref. 46 and 47.
Results
Characteristics of intermolecular π–π interactions
Our search for intermolecular π–π interactions revealed that 30 complexes (∼40% of the total set, Table 1) possessed at least one pair of interacting aromatic residues that satisfied the geometric restraints described in the Methods section. Therefore, the results described in this study correspond to these 31 complexes.
Table 1 Disordered protein dimeric complexes containing at least one aromatic pair
| PDB entry of the complex |
Chain ID of the IDP |
Name (IDP/binding partner) |
|
1a3b
|
I |
Boronate
inhibitor borolog 1/α-thrombin |
| 1apm |
I |
cAMP-dependent protein kinase inhibitor
PKI (5–24)/cAMP-dependent protein kinase |
| 1cee |
B |
Wiskott–Aldrich syndrome protein/Cdc42 |
| 1cmk |
I |
cAMP-dependent protein kinase inhibitor/cAMP-dependent protein kinase |
| 1dev |
B |
Smad anchor for receptor activation/SMAD2 MH2 domain |
| 1dow |
B |
β-Catenin/α-catenin |
| 1ej4 |
B |
Eukaryotic translation initiation factor 4E binding protein 1/eukariotic initiation factor 4E |
| 1fqj |
C |
Retinal rod rhodopsin-sensitive cGMP 3′,5′-cyclic phosphodiesterase γ-subunit/guanine nucleotide-binding protein |
|
1fv1
|
F |
Myelin basic protein/major histocompatibility complex |
|
1h26
|
E |
p53 (C-terminus)/cyclin A–CDK2 complex |
|
1i7x
|
B |
Epithetial cadherin/β-catenin |
| 1iwq |
B |
Myristoylated alanine-rich C-kinase substrate/calmodulin |
| 1jd5 |
B |
Cell death
protein
GRIM/apoptosis inhibitor DIAP1 |
| 1jsu |
C |
p27Kip1/cyclin A–CDK2 complex |
| 1kdx |
B |
pKID domain of the cAMP response element binding (CREB)/CREB binding protein |
|
1o9a
|
B |
Fibronectin binding
protein/fibronectin |
| 1ozs |
B |
Troponin I/troponin C |
|
1p16
|
D |
Phosphorylated peptide from the C-terminal of RNA polymerase II/mRNA capping enzyme α-subunit |
|
1p4b
|
P |
GCN4(7P14P), a peptide derived from the yeast transcription factor GCN4/antibody |
| 1pq1 |
B |
Isoform BIML of BCL2-like protein 11/Bcl-xL |
| 1rp3 |
B |
Anti-sigma factor FlgM/RNA polymerase sigma factor σ28 |
| 1ycq |
B |
p53 (N-terminus)/MDM2 |
|
2a6q
|
B |
Antitoxin yefM/toxin yoeB |
| 2cly |
C |
ATP synthase coupling factor 6/ATP synthase |
| 2iv8 |
P |
β-Arrestin 1/AP-2 |
| 2ivz |
E |
Colicin E9/TolB |
|
2o8a
|
I |
Protein phosphatase inhibitor 2/serine/threonine-protein phosphatase PP1-gamma catalytic subunit |
| 2pg1 |
I |
Cytoplasmic dynein 1 intermediate chain 2/dynein light chain |
| 2phe |
C |
α-trans-inducing protein/transcriptional coactivator PC4 |
| 4htc |
I |
Hirudin/α-thrombin |
We found a total of 83 aromatic residues distributed among IDPs and their binding partners; this number corresponds to aromatic residues that were unambiguously found to form π–π interactions in the complex interface. The observations indicate that phenylalanine often appears in intermolecular π–π interactions, as shown in Fig. 1 (blue bars). In substantially less proportion, tyrosine participates in π–π interactions at the interface of IDPs complexes (Fig. 1). Finally, tryptophan and histidine appear in very small proportions at the interfaces of complexes involving IDPs. As expected from the occurrence of aromatic amino acids at the interfaces of IDPs complexes, phenylalanine and tyrosine are the most preferred aromatic residues by IDPs to form π–π interactions (Fig. 1, red bars).
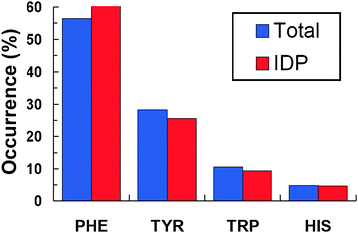 |
| | Fig. 1 Occurrence of aromatic residues involved in π–π interactions. The occurrence of each aromatic residue in both IDPs and their targets is labeled as “Total”, whereas the percentage of a particular aromatic residue that belongs to the IDP only is labeled as “IDP”. | |
We further looked into the prevalence of aromatic pairs that form π–π interactions (Fig. 2). As expected from the composition of the aromatic residues in the interfaces of the complexes, Phe-Phe and Phe-Tyr pairs account for more than 60% of the total formed π–π interactions involved in binding of IDPs. Interestingly, π–π interactions between tryptophan and phenylalanine or tyrosine also appear to be relatively populated at the interfaces of IDP complexes (Fig. 2). On the other hand, amino acid pairs composed by tryptophan and histidine are the least preferred by IDPs to form intermolecular π–π interactions (Fig. 2). Because the interface of complexes involving IDPs has a tendency to be hydrophobic, these results agree with previous observations showing that Phe-Tyr and Phe-Phe pairs are preferably formed in hydrophobic environments.49
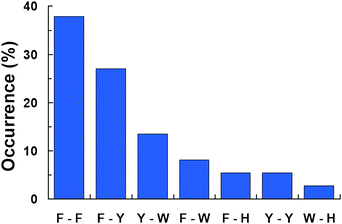 |
| | Fig. 2 Occurrence of aromatic pairs at the interface of the complexes. An aromatic pair was considered as described in the Methods section. For clarity, single letter codes were used for each aromatic amino acid (F = Phe; Y = Tyr; W = Trp; and H = His). | |
We also analyzed the preference for geometrical arrangement of aromatic–aromatic pairs in the 31 disordered protein complexes. We found that the T-shaped geometry is preferred over the stacking geometry for π–π interactions of IDPs (Fig. 3A). Interestingly, this preference for a T-shaped geometry extends to the most populated aromatic pairs (i.e., Phe-Phe and Phe-Tyr, Fig. 3B). These observations are particularly interesting because previous analyses of inter and intramolecular π–π interactions showed that stacking is preferred over the T-shaped geometry in globular proteins.13,14 On the other hand, potential of mean force of π–π interactions calculated using molecular dynamics simulations revealed that large Cα–Cα distances favor T-shaped π–π interactions. Thus, T-shaped geometries between aromatic residues may appear often in PPIs of IDPs because their interactions do not necessarily obey strict geometric complementarity.
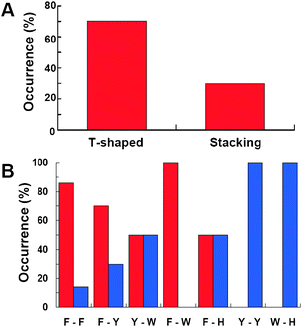 |
| | Fig. 3 Preference for geometrical arrangements of aromatic pairs. (A) Occurrence of T-shaped and stacking geometries of all aromatic pairs found at the interfaces of disordered protein complexes. (B) Occurrence of T-shaped (red) and stacking (blue) for specific aromatic pairs. Single letter codes were used for each aromatic amino acid (F = Phe; Y = Tyr; W = Trp; and H = His). | |
Secondary structure of aromatic residues
Secondary structure is an important feature that has been previously studied for the domains of IDPs that participate in binding. We calculated the secondary structure of each IDP in the 31 complexes studied here using STRIDE.50 This program recognizes secondary structural elements in proteins from their atomic coordinates and utilizes both hydrogen-bond energy and main-chain dihedral angles to define the secondary structure pattern. We further evaluated whether the backbone of aromatic residues involved in intermolecular π–π interactions posses a helix, a β-strand, a turn or a random coil conformation. We found that aromatic residues can populate any of the secondary structure motifs detected by STRIDE (Fig. 4). Nevertheless, we observed that 43% of the aromatic residues of IDP that participate in π–π interactions populate a helical backbone conformation, followed by a 34% of aromatic residues with random coil conformations. Only 23% of the aromatic residues involved in π–π interactions are found in either a β-strand or a turn.
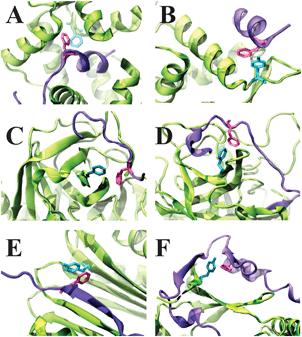 |
| | Fig. 4 Examples of the location of aromatic pairs at the interface of protein complexes based on the secondary structure. The complexes are: (A) the intrinsically disordered calmodulin-binding domain of a myristoylated alanine-rich C kinase substrate bound to calmodulin (PDB: 1iwq); (B) intrinsically disordered N-terminal region of p53 bound to MDM2 (PDB: 1ycq); (C) intrinsically disordered boronate inhibitor borolog 1 bound to α-thrombin (PDB: 1a3b); (D) hirudin bound to α-thrombin (PDB: 4htc); (E) cytoplasmic dynein 1 intermediate chain 2 bound to the dynein light chain (PDB 2pg1); (F) IDP p27kip1 bound to the cyclin dependent kinase 2-cyclin A complex (PDB: 1jsu). Protein structures are rendered as ribbons; IDPs are shown in violet, whereas their binding partners are shown in green. Aromatic residues are shown as sticks. | |
Although the backbone conformation in the bound state is important to understand the role of aromatic residues, understanding how these residues contribute to binding requires a characterization of the secondary structure of IDPs before binding. Therefore, we used the PSIPRED secondary structure prediction method51 and available experimental data to determine the secondary structure of the aromatic residues in the unbound IDPs. Based on the secondary structure prediction before (free IDP) and after (bound IDP) binding, we clustered aromatic residues in three groups: folding, non-folding and unfolding. The folding group includes residues whose backbone changes to a well-defined structural motif upon binding (i.e., random coil to helix). The non-folding group contains aromatic residues whose secondary structure does not change upon binding compared to the free form (i.e., coilfree → coilbound or helixfree → helix bound). Finally, the unfolding group includes residues whose secondary structure is lost upon binding (i.e., helix → coil). Surprisingly, we found that the majority of aromatic residues belong to the non-folding group, with 72% of the total aromatic residues. Only 25% of the aromatic residues were found in the folding group. Finally, only one residue, corresponding to the 3% of the total population, was found in the unfolding group.
Backbone mobility
It is well established that upon binding, IDPs do not necessarily undergo a complete disorder-to-order transition.52,53 As a result, backbone mobility of IDPs can vary in the bound complex, ranging from very rigid to fully flexible. This variation in flexibility can confer functional properties to different segments of IDPs in the bound state; for instance, residues with low mobility can work as bolts or anchors, conferring local structural stability; on the other hand, residues with high mobility can balance the enthalpy–entropy compensation in binding. To determine whether π–π interactions participate directly or indirectly in PPIs of IDPs, we calculated the relative backbone mobility of aromatic residues of IDPs involved as shown in Fig. 5. We found that ∼80% of the aromatic residues possess a relative backbone mobility of ≤0.5; in contrast, less than 5% of the aromatic residues displayed very high relative backbone mobility (i.e., values larger than 0.8). These results indicate that aromatic residues confer local rigidity to their backbone in the bound state. This phenomenon has been observed in the stabilization mechanism of a triosephosphate isomerase (TIM) homodimer. The formation and stability of the TIM homodimer require the disorder-to-order transition of two intrinsically disordered loops. Interestingly, experimental and computational studies showed that a structurally conserved aromatic residue of Plasmodium falciparumTIM (PfTIM), Tyr74, is essential for the stability of the homodimer.54–56 Tyr74, which forms π–π interactions at the interface of PfTIM, not only reduces the mobility of the interacting loops at the interface of the homodimer, but also contributes to the preservation of collective motions required for catalytic efficiency.56 Given the low backbone mobility of aromatic residues, it is possible that they not only contribute to the stability of IDPs in the bound state, but also translate this stability into function in the cell.
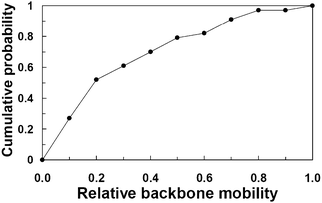 |
| | Fig. 5 Relative backbone mobility of the aromatic residues involved in π–π interactions. Relative mobility values (in arbitrary units) were calculated using the minimum and maximum crystallographic B-factors (for X-ray structures) or the root mean square fluctuation (for NMR structure) values of IDPs backbone atoms. Relative backbone mobility values were further normalized to a 0.0–1.0 scale. Values near or equal to 0 indicate low backbone mobility, whereas values near or equal to 1 indicate high backbone mobility. | |
Contribution of aromatic residues to the free energy of binding
To this point, the structural evidence suggests that π–π interactions play a role in stabilizing the protein–protein complexes of IDPs. For that reason, it is important to quantitatively evaluate the contribution of aromatic residues to the free energy of binding (ΔGbind). We used computational alanine scanning to determine whether or not aromatic residues act as hot spots, i.e., residues whose mutation to alanine produce an unfavorable change in binding free energy (ΔΔGbind) larger than 1 kcal mol−1. We found that, in general, most of the aromatic residues participating in π–π interactions satisfy this definition of hot spot, indicating that aromatic residues have a contribution to the free energy of binding (Fig. 6). Furthermore, analysis of the contribution of specific aromatic residues to ΔGbind revealed interesting information. First, more than 95% of the phenylalanine side chains of the IDPs have an important contribution to the ΔGbind of the complex, while 85% of the tyrosine side chains have a similar contribution to the ΔGbind. Second, the contribution to the free energy of binding is different between phenylalanine and tyrosine: while less than 15% of tyrosine side chains have ΔΔGbind values ≥ 3 kcal mol−1, 45% of phenylalanine side chains analyzed had similar ΔΔGbind values (Fig. 6). Third, despite the fact that tryptophan is not a preferred amino acid to form π–π interactions (Fig. 1 and 2), its contribution to ΔGbind is very large, with 80% of the tryptophan side chains having ΔΔGbind values larger than 4 kcal mol−1 (Fig. 6).
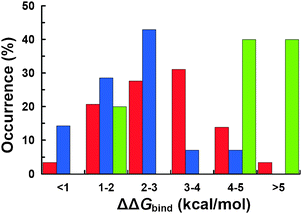 |
| | Fig. 6 Calculated changes in binding free energy (ΔΔGbind) for alanine mutations of IDPs involved in π–π interactions. We indicate the frequency of ΔΔGbind values for alanine mutations of phenylalanine (red), tyrosine (blue) and tryptophan (green). | |
Discussion
In this study we have established the importance of aromatic residues in PPIs of IDPs. Given the structural and thermodynamic characteristics of intermolecular interactions between aromatic residues, we propose that π–π interactions substantially contribute to binding for the following reasons: (a) π–π interactions facilitate specific binding of IDPs to their targets; for instance, molecular dynamics simulations have suggested that aromatic residues play an essential role in the specific binding of the intrinsically disordered transactivation domain of p53 and its regulator, the mouse double minute protein 2;57 (b) aromatic residues act as binding as ready-made recognition elements, i.e., because of the large proportion of non-folding residues, as described in the Results section; (c) given the preference of aromatic residues for self-association,39 π–π interactions can contribute to binding kinetics of IDPs; in this case, aromatic residues may increase the chances of formation of native-like encounter complexes, therefore decreasing the encounter times required for the fast binding of IDPs;58 (d) π–π interactions are an important source of stability to the bound complex of IDPs given their substantial contribution to the free energy of binding; and (e) given that PPIs of IDPs disobey strict geometric complementarity, T-shaped π–π interactions can act as effective molecular anchors at relatively long backbone distances without compromising the free energy of binding. Although aromatic residues appear to play an important role in PPIs involving IDPs, an important question arises from these observations: do aromatic residues link binding of IDPs and their function in the cell? To address this question, we analyzed the properties of π–π interactions and linked them to the function of IDPs. These IDPs and their function are discussed below.
β-Arrestin
β-Arrestins bind directly to the subunit β-1 of a heterotetrameric AP-2 adaptor complex using a conserved interaction motif, AspXXPheXXPheXXXArg.59–61 The AP-2 complex links many receptors to the clathrin endocytic machinery by binding to clathrin, dynamin and EPS-15, and is involved in the initiation of clathrin-coated pit formation.62 β-Arrestins recruit the AP-2 complex and clathrin, which target GPCRs for clathrin-mediated endocytosis. The crystal structure of β-arrestin 1 bound to AP-2 subunit β-161 revealed that two aromatic residues of β-arrestin 1, Phe388 and Phe391, interact with the hydrophobic groove on the AP-2 β-appendage via π–π interactions (Fig. 7A). We found that the relative backbone mobility of Phe388 and Phe391 is very small (<0.3); furthermore, we found that the binding ΔΔGbind is 3.9 kcal mol−1 for Phe388 and 2.3 kcal mol−1 for Phe391, in agreement with experiments showing that mutations to alanine of both phenylalanines prevent AP-2 from binding to GST-β-arrestin C1.61 Of particular interest is the highly conserved Phe391, which interacts via π–π interactions with Trp841 from AP-2. Because (a) Trp841 is a highly conserved residue and (b) the contribution of Phe391 to the ΔGbind is very large, it is likely that the interaction between Phe391 and Trp841 is essential for the stability of the β-arrestin–AP-2 complex, and therefore for clathrin-coated vesicle assembly.
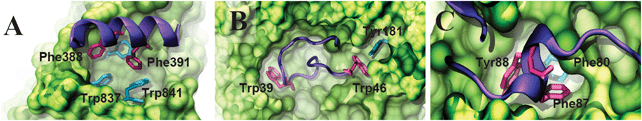 |
| | Fig. 7 Examples of molecular complexes solved by X-ray crystallography in which aromatic residues link binding with function. (A) β-Arrestin 1 bound to AP-2 subunit β-1 (PDB: 2iv8); (B) intrinsically disordered region of colicin E9 bound to TolB (PDB: 2ivz); (C) p27kip1 bound to the cyclin dependent kinase 2–cyclin A complex (PDB: 1jsu). IDPs are shown as violet ribbons, and their binding partners are rendered as green surfaces. Aromatic residues discussed in the main text are shown as sticks. | |
Colicin E9
Colicins are protein antibiotics produced mostly by Gram-negative microorganisms. Colicins specifically target E. coli, and typically kill cells by depolarizing the inner membrane or degrading nucleic acid in the cytoplasm.63 Colicins exert their cytotoxic effect by binding an outer membrane receptor and subsequently translocating into the cellviaTol or Ton proteins within the periplasm.64 The intrinsically disordered translocation domain of E group colicins recruits TolB in the periplasm of E. coli, leading to toxin translocation.65 Experimental studies revealed that the intrinsically disordered region of colicin E9 uses two tryptophans for binding to TolB with sub-micromolar affinity (Fig. 7B).65 Inspection of the crystal structure revealed that one of these tryptophans, Trp46, interacts via T-shaped π–π interactions with Tyr181 of TolB (Fig. 7B). Alanine mutation of Trp46 showed that this residue has a very large contribution to ΔGbind, with ΔΔGbind = 7 kcal mol−1. This observation is in agreement with experiments showing that mutant Trp46Ala prevents colicin E9 from binding to TolB in vivo.66 Although these observations indicate that Trp46 directly participates in the stability of the colicin E9–TolB complex, we suggest that this residue plays a key role in “fly casting”67 TolB. The dual role of Trp46 allows the intrinsically disordered N-terminus of colicin E9 to perform a specific “blind catch” of TolB from the periplasm, and to provide a sufficiently strong intermolecular interaction with TolB for import into the cell.
Tumor suppressor p27kip1
Tumor suppressor p27kip1 is a cell-cycle regulatory protein that blocks progression from G1 to S phase of the cell division cycle by binding to the cyclin dependent kinase 2 (CDK2)–cyclin A complex.68,69 p27kip1 is an intrinsically disordered protein that undergoes coupled folding and binding upon binding with high affinity to the CDK2–cyclin A complex.69,70 Crystallographic studies have shown that p27kip1 exerts its biological activity by occluding the ATP-binding site of CDK2, and therefore preventing ATP from binding.69 The crystal structure of the complex showed that upon binding, the region of p27kip1 that occludes the ATP-binding undergoes a disorder-to-order transition, forming a short 310 helix (Fig. 7C). In the complex, a highly conserved aromatic residue of p27kip1 (Tyr88) possesses a primary role in inhibiting ATP binding by mimicking the stabilizing electrostatic interactions observed in the native ATP–CDK2 complex. Interestingly, we found a π–π interaction between p27kip1 and the ATP-binding site of CDK2; this interaction involves conserved Phe87 of p27kip1 and Phe80 of CDK2 (Fig. 7C). Phe87 possesses several properties that correlate with its role in the stability of the complex: (a) its side chain becomes largely buried upon binding, with a change in solvent-accessible surface area (ΔSASA) of 122 Å2 (75% of the accessible surface); (b) its relative backbone mobility is very small (∼0.1); and (c) its contribution to the free energy of binding is large, with a ΔΔGbind value of 3 kcal mol−1. Based on these observations, we suggest that Phe87 restrains the position of Tyr88 and the backbone of p27kip1 at the ATP-binding site. As a result, Phe87 ensures that key electrostatic interactions occurring between ATP and CDK2 are correctly mimicked by Tyr8869 and therefore allowing p27kip1 to effectively block ATP binding, which is a requirement to inhibit cell proliferation.
Wiskott–Aldrich syndrome protein
The Rho-family GTP-hydrolyzing proteins (GTPases), Cdc42, Rac and Rho, act as molecular switches in signaling pathways that regulate cytoskeletal architecture, gene expression and progression of the cell cycle.71Cdc42 and Rac transmit many signals through GTP-dependent binding to effector proteins containing a Cdc42/Rac-interactive-binding motif. One such effector, the Wiskott–Aldrich syndrome protein (WASP), is postulated to link activation of Cdc42 directly to the rearrangement of actin via the Arp2/3 complex.72,73 It has also been demonstrated that the Cdc42–WASP interaction is critical for stromal cell-derived factor 1-induced chemotaxis of T cells.74 Nuclear magnetic resonance (NMR) experiments revealed that WASP interacts with Cdc42 by using a coil–β-strand–hairpin–helix fold (Fig. 8A).75 In the NMR structures, we found an interaction between two aromatic residues: Trp252 from WASP and Tyr64 from Cdc42. Trp252 is located at the N-terminus of the hairpin (residues Gly251-Leu262) (Fig. 8A). The NMR structure of the autoinhibited form of WASP76 showed that Trp252 has π–π contacts with another aromatic residue of WASP, Tyr279 (Fig. S1, ESI‡). Surprisingly, a structural comparison of the region Trp252-Asp259 showed an almost identical conformation between bound and autoinhibited WASP (Fig. S2, ESI‡). Even more surprising is the fact that the position of the side chain of Trp252 is highly restrained in both bound and autoinhibited WASP. Based on these observations, we suggest that Trp252 contributes to the formation of the Cdc42–WASP complex by restraining the mobility of region Trp252-Asp259. As a result, Trp252 helps Trp252-Asp259 to preserve its bound-like conformation, which may act as a preformed recognition motif that provides binding specificity for the fast Cdc42–WASP association necessary for the temporal regulation and integration of WASP-mediated cellular responses.77
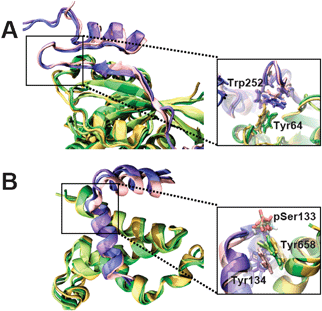 |
| | Fig. 8 Examples of molecular complexes solved by NMR in which aromatic residues link binding with function. (A) Wiskott–Aldrich syndrome protein bound to Cdc42 (PDB: 1cee); (B) phosphorylated kinase inducible domain of cAMP response-element binding protein (CREB) bound to the CREB binding protein (PDB: 1kdx). IDPs are shown as pink/blue/violet ribbons. The black squares highlight the regions where aromatic residues that link binding and function are found. Aromatic residues discussed in the text are rendered as sticks. | |
cAMP response-element binding protein
The cAMP-response element binding protein (CREB) is a target of signaling pathways activated by a diverse array of stimuli.78 In response to these stimuli, the kinase inducible domain (KID) of CREB is phosphorylated at residue Ser133 by protein kinase A.79 Phosphorylated CREB subsequently activates transcription by recruiting and associating with the transcriptional co-activator, CREB binding protein (CBP),79,80 which results in the recruitment and stabilization of the RNA polymerase II transcription complex at the TATA box. Additionally, CBP also contributes to CREB-mediated transcription by affecting the chromatin structure.78NMR studies have shown that upon binding, the intrinsically disordered phosphorylated KID (pKID) domain of CREB folds as two α-helices linked by a short loop (Fig. 8B).81,82 Although it has been suggested that phosphorylated Ser133 is essential for the stability of the bound complex,83 a computational study showed that a neighboring residue, Tyr134, plays a direct role in binding specificity through a mechanism that relies on thermodynamic control.48 Tyr134, which is absolutely conserved amongst CREB homologues, interacts with the aromatic residue Tyr658 of mouse CBP (Fig. 8B). Mutation of Tyr134 to alanine showed that this residue has a substantial contribution to the free energy of binding (ΔΔGbind ≥ 2 kcal mol−1),48 in agreement with experiments showing that Tyr134 → Ala134 mutation blocks association of pKID with CBP in vitro.84 Furthermore, we found that Tyr134 appears to restrain the side chain mobility of Tyr658, which facilitates the formation of a hydrogen bond between the hydroxyl moiety of Tyr685 and phosphorylated Ser133. We suggest that Tyr134 has an essential role in the CREB-mediated transcription activation by acting as a hub that links binding specificity and stability of the CREB–CBP complex, which is essential to promote recruitment of the transcriptional apparatus.
The intrinsically disordered BCL2-like protein 11 (BCL2L11) is a member of the Bcl-2 family of proteins that either inhibit or activate apoptosis.85BCL2L11 belongs to the group of Bcl-2 proteins which induces death of the T cells following αβTCR activation by interacting with Bcl-xL, another member of the BCL-2 family.86 Because of the biological relevance of the complex, Bcl-xL is a very attractive target for cancer therapy with potential use in treating lymphoid malignancies, small-cell lung cancer and chronic lymphocytic leukemia.87 Crystallographic studies revealed that the BH3 region of isoform BIML of mouse BCL2L11 binds to Bcl-xL using a long α-helix spanning residues Pro86-Asp113 (Fig. 9).88 We used the PONDR-FIT server89 to perform a prediction of intrinsic disorder of full-length BIML using its amino acid sequence. The prediction indicates that although ∼65% of BIML is intrinsically disordered, the helix that binds Bcl-xL is likely to preexist in solution (Fig. 9). We found a T-shaped π–π interaction at the interface of the complex between Phe101 (BIML) and Phe97 (Bcl-xL); this interaction is shown in Fig. 9. Computational alanine scanning indicated that the contribution of conserved Phe101 to the free energy of binding is very large (ΔΔGbind = 4 kcal mol−1). Furthermore, we found a particularly interesting structural feature: Phe101 participates in an array of interactions involving aromatic clusters with Tyr105 of BIML and Phe97 of Bcl-xL. Strikingly, these aromatic residues are located at positions i, i + 4 and i, i − 4 relative to Phe101. Because aromatic residues at this positions have been shown to contribute to the stability of α-helices,90 we suggest that Phe101 links binding with function via two mechanisms: (a) Phe101 acts as an anchor residue because of its large contribution to the ΔGbind and a ΔSASA of 122 Å2 (or ∼80% of the accessible surface) and (b) the formation of the π–π interaction network Phe97Bcl-xL-Phe101BIML-Tyr105BIML is probably a fingerprint required for specific binding of BIML to Bcl-xL, which is necessary for the induction of T-cell apoptosis.
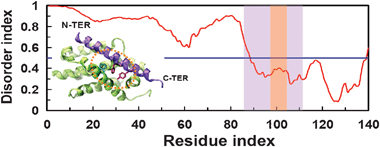 |
| | Fig. 9 Disorder prediction of the isoform BIML of mouse BCL2-like protein 11. We show the crystallographic structure of BIML (violet) bound to Bcl-xL (green). The PDB accession code of the complex is 1pq1. In the disorder index plot, the region of BIML that binds to Bcl-xL is highlighted in pale violet; the region of BIML that participates in the π–π interaction network Phe97Bcl-xL-Phe101BIML-Tyr105BIML (sticks) is highlighted in pale orange. | |
Conclusion
Analysis of available three-dimensional structures of protein–protein complexes involving intrinsically disordered proteins showed that aromatic–aromatic intermolecular interactions occur frequently at the interface of these complexes. Structural and thermodynamic analysis revealed that aromatic residues substantially contribute to the binding of IDPs. Furthermore, we found that aromatic residues work as ready-made recognition motifs for specific binding, anchor residues91 and conformational locks. By using available experimental data for several IDPs, we found that aromatic residues not only play an important role in PPIs of IDPs, but also connect binding with their function in the cell. This study paves the way to understand in atomic detail the inner workings of IDPs interaction networks and their connection with biological functions, with potential applications in drug design.
Acknowledgements
The author is grateful to A. Keith Dunker and Pedro Romero for fruitful discussions. L.M.E.-F. was supported by the American Heart Association and the Minnesota Supercomputing Institute at the University of Minnesota.
References
-
B. Alberts, Molecular Biology of the Cell, Garland Science, New York, 2002 Search PubMed.
- D. Reichmann,
et al., The modular architecture of protein–protein binding interfaces, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 57–62 CrossRef CAS.
- L. Lo Conte, C. Chothia and J. Janin, The atomic structure of protein–protein recognition sites, J. Mol. Biol., 1999, 285, 2177–2198 CrossRef CAS.
- S. Jones and J. M. Thornton, Principles of protein–protein interactions, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 13–20 CrossRef CAS.
- J. Janin and C. Chothia, The structure of protein–protein recognition sites, J. Biol. Chem., 1990, 265, 16027–16030 CAS.
- A. A. Bogan and K. S. Thorn, Anatomy of hot spots in protein interfaces, J. Mol. Biol., 1998, 280, 1–9 CrossRef CAS.
- J. Janin, Principles of protein–protein recognition from structure to thermodynamics, Biochimie, 1995, 77, 497–505 CrossRef CAS.
- O. Keskin, A. Gursoy, B. Ma and R. Nussinov, Principles of protein–protein interactions: what are the preferred ways for proteins to interact?, Chem. Rev., 2008, 108, 1225–1244 CrossRef CAS.
- O. Keskin, B. Ma, K. Rogale, K. Gunasekaran and R. Nussinov, Protein–protein interactions: organization, cooperativity and mapping in a bottom-up Systems Biology approach, Phys. Biol., 2005, 2, S24–S35 CrossRef CAS.
- M. M. Gromiha, K. Yokota and K. Fukui, Energy based approach for understanding the recognition mechanism in protein–protein complexes, Mol. Biosyst., 2009, 5, 1779–1786 RSC.
- S. K. Burley and G. A. Petsko, Aromatic–aromatic interaction: a mechanism of protein structure stabilization, Science, 1985, 229, 23–28 CAS.
- S. K. Burley and G. A. Petsko, Amino–aromatic interactions in proteins, FEBS Lett., 1986, 203, 139–143 CrossRef CAS.
- G. B. McGaughey, M. Gagne and A. K. Rappe, pi-Stacking interactions. Alive and well in proteins, J. Biol. Chem., 1998, 273, 15458–15463 CrossRef CAS.
- K. I. Cho, K. Lee, K. H. Lee, D. Kim and D. Lee, Specificity of molecular interactions in transient protein–protein interaction interfaces, Proteins: Struct., Funct., Bioinf., 2006, 65, 593–606 CrossRef CAS.
- V. N. Uversky, J. R. Gillespie and A. L. Fink, Why are “natively unfolded” proteins unstructured under physiologic conditions?, Proteins: Struct., Funct., Genet., 2000, 41, 415–427 CrossRef CAS.
- V. N. Uversky, Natively unfolded proteins: a point where biology waits for physics, Protein Sci., 2002, 11, 739–756 CrossRef CAS.
- P. Tompa, Intrinsically unstructured proteins, Trends Biochem. Sci., 2002, 27, 527–533 CrossRef CAS.
- P. E. Wright and H. J. Dyson, Intrinsically unstructured proteins: re-assessing the protein structure–function paradigm, J. Mol. Biol., 1999, 293, 321–331 CrossRef CAS.
- A. K. Dunker and Z. Obradovic, The protein trinity—linking function and disorder, Nat. Biotechnol., 2001, 19, 805–806 CrossRef CAS.
- P. Tompa, The interplay between structure and function in intrinsically unstructured proteins, FEBS Lett., 2005, 579, 3346–3354 CrossRef CAS.
- P. Tompa, C. Szasz and L. Buday, Structural disorder throws new light on moonlighting, Trends Biochem. Sci., 2005, 30, 484–489 CrossRef CAS.
- V. N. Uversky, C. J. Oldfield and A. K. Dunker, Showing your ID: intrinsic disorder as an ID for recognition, regulation and cell signaling, J. Mol. Recognit., 2005, 18, 343–384 CrossRef CAS.
- A. K. Dunker, I. Silman, V. N. Uversky and J. L. Sussman, Function and structure of inherently disordered proteins, Curr. Opin. Struct. Biol., 2008, 18, 756–764 CrossRef CAS.
- H. J. Dyson and P. E. Wright, Intrinsically unstructured proteins and their functions, Nat. Rev. Mol. Cell Biol., 2005, 6, 197–208 CrossRef CAS.
- V. N. Uversky and A. K. Dunker, Understanding protein non-folding, Biochim. Biophys. Acta, 2010, 1804, 1231–1264 CAS.
- L. M. Espinoza-Fonseca, Leucine-rich hydrophobic clusters promote folding of the N-terminus of the intrinsically disordered transactivation domain of p53, FEBS Lett., 2009, 583, 556–560 CrossRef CAS.
- K. P. Wu and J. Baum, Detection of transient interchain interactions in the intrinsically disordered protein alpha-synuclein by NMR paramagnetic relaxation enhancement, J. Am. Chem. Soc., 2010, 132, 5546–5547 CrossRef CAS.
- D. F. Lowry, A. Stancik, R. M. Shrestha and G. W. Daughdrill, Modeling the accessible conformations of the intrinsically unstructured transactivation domain of p53, Proteins: Struct., Funct., Bioinf., 2008, 71, 587–598 CrossRef CAS.
- F. Huang,
et al., Multiple conformations of full-length p53 detected with single-molecule fluorescence resonance energy transfer, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 20758–20763 CrossRef CAS.
- K. Xiong,
et al., Direct Observations of Conformational Distributions of Intrinsically Disordered p53 Peptides Using UV Raman and Explicit Solvent Simulations, J. Phys. Chem. A, 2011 DOI:10.1021/jp112235d.
- C. J. Oldfield,
et al., Coupled folding and binding with alpha-helix-forming molecular recognition elements, Biochemistry, 2005, 44, 12454–12470 CrossRef CAS.
- V. Csizmok,
et al., Primary contact sites in intrinsically unstructured proteins: the case of calpastatin and microtubule-associated protein 2, Biochemistry, 2005, 44, 3955–3964 CrossRef CAS.
- M. Fuxreiter, I. Simon, P. Friedrich and P. Tompa, Preformed structural elements feature in partner recognition by intrinsically unstructured proteins, J. Mol. Biol., 2004, 338, 1015–1026 CrossRef CAS.
- A. Mohan,
et al., Analysis of molecular recognition features (MoRFs), J. Mol. Biol., 2006, 362, 1043–1059 CrossRef CAS.
- B. Mészáros, P. Tompa, I. Simon and Z. Dosztányi, Molecular principles of the interactions of disordered proteins, J. Mol. Biol., 2007, 372, 549–561 CrossRef.
- B. Mészáros, I. Simon and Z. Dosztányi, The expanding view of protein–protein interactions: complexes involving intrinsically disordered proteins, Phys. Biol., 2011, 8, 035003 CrossRef.
- B. Mészáros, I. Simon and Z. Dosztányi, Prediction of protein binding regions in disordered proteins, PLoS Comput. Biol., 2009, 5, e1000376 Search PubMed.
- C. J. Brown, A. K. Johnson and G. W. Daughdrill, Comparing models of evolution for ordered and disordered proteins, Mol. Biol. Evol., 2010, 27, 609–621 CrossRef CAS.
- M. L. Waters, Aromatic interactions in model systems, Curr. Opin. Chem. Biol., 2002, 6, 736–741 CrossRef CAS.
- C. D. Tatko and M. L. Waters, Selective aromatic interactions in beta-hairpin peptides, J. Am. Chem. Soc., 2002, 124, 9372–9373 CrossRef CAS.
- Y. Cheng,
et al., Mining alpha-helix-forming molecular recognition features with cross species sequence alignments, Biochemistry, 2007, 46, 13468–13477 CrossRef CAS.
- P. Romero,
et al., Sequence complexity of disordered protein, Proteins: Struct., Funct., Genet., 2001, 42, 38–48 CrossRef CAS.
- H. M. Berman,
et al., The Protein Data Bank, Nucleic Acids Res., 2000, 28, 235–242 CrossRef CAS.
- J. C. Phillips,
et al., Scalable molecular dynamics with NAMD, J. Comput. Chem., 2005, 26, 1781–1802 CrossRef CAS.
- W. Humphrey, A. Dalke and K. Schulten, VMD: visual molecular dynamics, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS , 27–28.
- T. Kortemme and D. Baker, A simple physical model for binding energy hot spots in protein–protein complexes, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 14116–14121 CrossRef CAS.
- T. Kortemme, D. E. Kim and D. Baker, Computational alanine scanning of protein–protein interfaces, Sci. STKE, 2004, 2004, pl2 CrossRef.
- L. M. Espinoza-Fonseca, Thermodynamic aspects of coupled binding and folding of an intrinsically disordered protein: a computational alanine scanning study, Biochemistry, 2009, 48, 11332–11334 CrossRef CAS.
- R. Chelli, F. L. Gervasio, P. Procacci and V. Schettino, Stacking and T-shape competition in aromatic–aromatic amino acid interactions, J. Am. Chem. Soc., 2002, 124, 6133–6143 CrossRef CAS.
- D. Frishman and P. Argos, Knowledge-based protein secondary structure assignment, Proteins: Struct., Funct., Genet., 1995, 23, 566–579 CrossRef CAS.
- D. T. Jones, Protein secondary structure prediction based on position-specific scoring matrices, J. Mol. Biol., 1999, 292, 195–202 CrossRef CAS.
- P. Tompa and M. Fuxreiter, Fuzzy complexes: polymorphism and structural disorder in protein–protein interactions, Trends Biochem. Sci., 2008, 33, 2–8 CrossRef CAS.
- A. B. Sigalov, A. V. Zhuravleva and V. Y. Orekhov, Binding of intrinsically disordered proteins is not necessarily accompanied by a structural transition to a folded form, Biochimie, 2007, 89, 419–421 CrossRef CAS.
- K. Maithal,
et al., Subunit interface mutation disrupting an aromatic cluster in Plasmodium falciparum triosephosphate isomerase: effect on dimer stability, Protein Eng., 2002, 15, 575–584 CrossRef CAS.
- B. Gopal,
et al., Cavity-creating mutation at the dimer interface of Plasmodium falciparum triosephosphate isomerase: restoration of stability by disulfide cross-linking of subunits, Biochemistry, 1999, 38, 478–486 CrossRef CAS.
- L. M. Espinoza-Fonseca, C. Wong-Ramirez and J. G. Trujillo-Ferrara, Tyr74 is essential for the formation, stability and function of Plasmodium falciparum triosephosphate isomerase dimer, Arch. Biochem. Biophys., 2010, 494, 46–57 CrossRef CAS.
- L. M. Espinoza-Fonseca and J. Garcia-Machorro, Aromatic–aromatic interactions in the formation of the MDM2–p53 complex, Biochem. Biophys. Res. Commun., 2008, 370, 547–551 CrossRef CAS.
- Y. Huang and Z. Liu, Kinetic advantage of intrinsically disordered proteins in coupled folding-binding process: a critical assessment of the “fly-casting” mechanism, J. Mol. Biol., 2009, 393, 1143–1159 CrossRef CAS.
- S. A. Laporte,
et al., The beta2-adrenergic receptor/betaarrestin complex recruits the clathrin adaptor AP-2 during endocytosis, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 3712–3717 CrossRef CAS.
- S. A. Laporte, R. H. Oakley, J. A. Holt, L. S. Barak and M. G. Caron, The interaction of beta-arrestin with the AP-2 adaptor is required for the clustering of beta 2-adrenergic receptor into clathrin-coated pits, J. Biol. Chem., 2000, 275, 23120–23126 CrossRef CAS.
- E. M. Schmid,
et al., Role of the AP2 beta-appendage hub in recruiting partners for clathrin-coated vesicle assembly, PLoS Biol., 2006, 4, e262 Search PubMed.
- T. Kirchhausen, Adaptors for clathrin-mediated traffic, Annu. Rev. Cell Dev. Biol., 1999, 15, 705–732 CrossRef CAS.
- I. Brook, Bacterial interference, Crit. Rev. Microbiol., 1999, 25, 155–172 CrossRef CAS.
- C. J. Lazdunski,
et al., Colicin import into Escherichia coli cells, J. Bacteriol., 1998, 180, 4993–5002 CAS.
- S. R. Loftus,
et al., Competitive recruitment of the periplasmic translocation portal TolB by a natively disordered domain of colicin E9, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 12353–12358 CrossRef CAS.
- S. L. Hands,
et al., Interactions of TolB with the translocation domain of colicin E9 require an extended TolB box, J. Bacteriol., 2005, 187, 6733–6741 CrossRef CAS.
- B. A. Shoemaker, J. J. Portman and P. G. Wolynes, Speeding molecular recognition by using the folding funnel: the fly-casting mechanism, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 8868–8873 CrossRef CAS.
- E. R. Lacy,
et al., p27 binds cyclin–CDK complexes through a sequential mechanism involving binding-induced protein folding, Nat. Struct. Mol. Biol., 2004, 11, 358–364 CAS.
- A. A. Russo, P. D. Jeffrey, A. K. Patten, J. Massague and N. P. Pavletich, Crystal structure of the p27Kip1 cyclin-dependent-kinase inhibitor bound to the cyclin A–Cdk2 complex, Nature, 1996, 382, 325–331 CrossRef CAS.
- G. M. Verkhivker,
et al., Simulating disorder–order transitions in molecular recognition of unstructured proteins: where folding meets binding, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 5148–5153 CrossRef CAS.
- A. Hall, Rho GTPases and the actin cytoskeleton, Science, 1998, 279, 509–514 CrossRef CAS.
- M. Symons,
et al., Wiskott–Aldrich syndrome protein, a novel effector for the GTPase CDC42Hs, is implicated in actin polymerization, Cell, 1996, 84, 723–734 CrossRef CAS.
- T. H. Millard, S. J. Sharp and L. M. Machesky, Signalling to actin assembly via the WASP (Wiskott–Aldrich syndrome protein)-family proteins and the Arp2/3 complex, Biochem. J., 2004, 380, 1–17 CrossRef CAS.
- E. Haddad,
et al., The interaction between Cdc42 and WASP is required for SDF-1-induced T-lymphocyte chemotaxis, Blood, 2001, 97, 33–38 CrossRef CAS.
- N. Abdul-Manan,
et al., Structure of Cdc42 in complex with the GTPase-binding domain of the ‘Wiskott–Aldrich syndrome’ protein, Nature, 1999, 399, 379–383 CrossRef CAS.
- A. S. Kim, L. T. Kakalis, N. Abdul-Manan, G. A. Liu and M. K. Rosen, Autoinhibition and activation mechanisms of the Wiskott–Aldrich syndrome protein, Nature, 2000, 404, 151–158 CrossRef CAS.
- L. Hemsath, R. Dvorsky, D. Fiegen, M. F. Carlier and M. R. Ahmadian, An electrostatic steering mechanism of Cdc42 recognition by Wiskott–Aldrich syndrome proteins, Mol. Cell, 2005, 20, 313–324 CrossRef CAS.
- A. J. Shaywitz and M. E. Greenberg, CREB: a stimulus-induced transcription factor activated by a diverse array of extracellular signals, Annu. Rev. Biochem., 1999, 68, 821–861 CrossRef CAS.
- J. C. Chrivia,
et al., Phosphorylated CREB binds specifically to the nuclear protein CBP, Nature, 1993, 365, 855–859 CrossRef CAS.
- R. P. Kwok,
et al., Nuclear protein CBP is a coactivator for the transcription factor CREB, Nature, 1994, 370, 223–226 CrossRef CAS.
- I. Radhakrishnan,
et al., Solution structure of the KIX domain of CBP bound to the transactivation domain of CREB: a model for activator
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :coactivator interactions, Cell, 1997, 91, 741–752 CrossRef CAS.
:coactivator interactions, Cell, 1997, 91, 741–752 CrossRef CAS.
- K. Sugase, H. J. Dyson and P. E. Wright, Mechanism of coupled folding and binding of an intrinsically disordered protein, Nature, 2007, 447, 1021–1025 CrossRef CAS.
- T. Zor, B. M. Mayr, H. J. Dyson, M. R. Montminy and P. E. Wright, Roles of phosphorylation and helix propensity in the binding of the KIX domain of CREB-binding protein by constitutive (c-Myb) and inducible (CREB) activators, J. Biol. Chem., 2002, 277, 42241–42248 CrossRef CAS.
- D. Parker,
et al., Analysis of an activator:coactivator complex reveals an essential role for secondary structure in transcriptional activation, Mol. Cell, 1998, 2, 353–359 CrossRef CAS.
- J. M. Adams and S. Cory, The Bcl-2 protein family: arbiters of cell survival, Science, 1998, 281, 1322–1326 CrossRef CAS.
- P. Bouillet,
et al., BH3-only Bcl-2 family member Bim is required for apoptosis of autoreactive thymocytes, Nature, 2002, 415, 922–926 CrossRef CAS.
- M. Vogler, D. Dinsdale, M. J. Dyer and G. M. Cohen, Bcl-2 inhibitors: small molecules with a big impact on cancer therapy, Cell Death Differ., 2009, 16, 360–367 CrossRef CAS.
- X. Liu, S. Dai, Y. Zhu, P. Marrack and J. W. Kappler, The structure of a Bcl-xL/Bim fragment complex: implications for Bim function, Immunity, 2003, 19, 341–352 CrossRef CAS.
- B. Xue, R. L. Dunbrack, R. W. Williams, A. K. Dunker and V. N. Uversky, PONDR-FIT: a meta-predictor of intrinsically disordered amino acids, Biochim. Biophys. Acta, 2010, 1804, 996–1010 CAS.
- S. Aravinda,
et al., Aromatic–aromatic interactions in crystal structures of helical peptide scaffolds containing projecting phenylalanine residues, J. Am. Chem. Soc., 2003, 125, 5308–53015 CrossRef CAS.
- D. Rajamani, S. Thiel, S. Vajda and C. J. Camacho, Anchor residues in protein–protein interactions, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 11287–11292 CrossRef CAS.
Footnotes |
| † Published as part of a Molecular BioSystems themed issue on Intrinsically Disordered Proteins: Guest Editor M. Madan Babu. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c1mb05239j |
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here.