Protein intrinsic disorder and induced pluripotent stem cells†‡
Bin
Xue
ab,
Christopher J.
Oldfield
b,
Ya-Yue
Van
c,
A. Keith
Dunker
*b and
Vladimir N.
Uversky
*abd
aDepartment of Molecular Medicine, College of Medicine, University of South Florida, Tampa, Florida 33612, USA. E-mail: bxue@health.usf.edu
bCenter for Computational Biology and Bioinformatics, Indiana University School of Medicine, Indianapolis, IN 46202, USA. E-mail: cjoldfie@umail.iu.edu; kedunker@iupui.edu
cMolecular Kinetics Inc., Indianapolis, IN 46254, USA. E-mail: main@molecularkinetics.com
dInstitute for Biological Instrumentation, Russian Academy of Sciences, 142290 Pushchino, Moscow Region, Russia. E-mail: vuversky@health.usf.edu
First published on 14th July 2011
Abstract
Induced pluripotent stem (iPS) cells can be obtained from terminally differentiated somatic cells by overexpression of defined sets of reprogramming transcription factors. These protein sets have been called the Yamanaka factors, namely Sox2, Oct3/4 (Pou5f1), Klf4, and c-Myc, and the Thomson factors, namely Sox2, Oct3, Lin28, and Nanog. Other sets of proteins, while not essential for the formation of iPS cells, are important for improving the efficiency of the induction and still other sets of proteins are important as markers for embryonic stem cells. Structural information about most of these important proteins is very sparse. Our bioinformatics analysis herein reveals that these reprogramming factors and most of the efficiency-improving and embryonic stem cell markers are highly enriched in intrinsic disorder. As is typical for transcription factors, these proteins are modular. Specific sites for interaction with other proteins and DNA are dispersed in the long regions of intrinsic disorder. These highly dynamic interaction sites are evidently responsible for the delicate interplay among various molecules. The bioinformatics analysis given herein should facilitate the investigation of the roles and organization of these modular interaction sites, thereby helping to shed further light on the pathways that underlie the mechanism(s) by which terminally differentiated cells are converted to iPS cells.
Introduction
Cells are the fundamental units of life. In mammals, cells have a hierarchical classification based on their cellular differentiation at various developmental stages. In terms of developmental potency, these cells can be categorized into four classes: totipotent, pluripotent, multipotent, and unipotent.1 As indicated by the names, totipotent cells have the ability to differentiate into all other cell types. Only zygotes and blastomeres at the earliest developmental stage of natural or cloned embryos are totipotent cells. Pluripotent cells are capable of differentiating into all other cell types except the trophoblast lineage. There are currently five types of pluripotent cells:![[E with combining low line]](https://www.rsc.org/images/entities/char_0045_0332.gif) mbryonic
mbryonic ![[S with combining low line]](https://www.rsc.org/images/entities/char_0053_0332.gif) tem (ES) cells, mbryonic
tem (ES) cells, mbryonic ![[G with combining low line]](https://www.rsc.org/images/entities/char_0047_0332.gif) erm cells (EGCs), mbryonic
erm cells (EGCs), mbryonic ![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) arcinoma cells (ECCs),
arcinoma cells (ECCs), ![[i with combining low line]](https://www.rsc.org/images/entities/char_0069_0332.gif) nduced
nduced ![[P with combining low line]](https://www.rsc.org/images/entities/char_0050_0332.gif) luripotent tem (iPS) cells, and
luripotent tem (iPS) cells, and ![[m with combining low line]](https://www.rsc.org/images/entities/char_006d_0332.gif) ulti-potent ermline tem (mGS) cells.2–6Multipotent cells are able to differentiate along multiple cell lineages within the same tissue. Unipotent cells can only differentiate into one specific cell type. Apparently, the greater the differentiation ability, the more complicated the signaling and regulation inside the cell.
ulti-potent ermline tem (mGS) cells.2–6Multipotent cells are able to differentiate along multiple cell lineages within the same tissue. Unipotent cells can only differentiate into one specific cell type. Apparently, the greater the differentiation ability, the more complicated the signaling and regulation inside the cell.
In these different cell types, pluripotent cells have attracted a lot of attention over the last several years. Among the five different sub-classes of pluripotent cells, iPS cells show many unique features, such as the ability of being differentiated into many other types of cells and being cultured independently. In addition, (iPS) cells can be generated from patients with a variety of genetic diseases. These disease-specific iPS cells offer a unique opportunity to recapitulate both normal and pathologic human tissue formation in vitro and therefore open new and unique ways for understanding disease mechanisms, drug screening, and toxicology.7 iPS cells have also been used in regenerative medicine and other medical treatment without causing any immunologic rejection or other ethical concerns.8,9 Hence, the technologies for inducing iPS cells from somatic cells have opened up a brand new field, connecting basic research to personalized medicine.
In recent efforts to obtain iPS cells, specific combinations of transcription factors were shown to be able to induce the transformation of various types of terminally differentiated somatic cells to iPS cells. In 2006, Takahashi and Yamanaka obtained mouse iPS cells from embryonic and fibroblast cells by overexpressing Sox2, Oct3/4 (Pou5f1), Klf4, and c-Myc.8 Later on, they also produced human iPS cells from fibroblast by using the same set of human transcription factors.9 During the same period of time, Thomson's group accomplished the transformation of human somatic cells into iPS cells by overexpressing a different set of four factors: Sox2, Oct3, Lin28, and Nanog.10 These two sets of proteins were later called Yamanaka factors and Thomson factors,11 or SOKM and SOLN,12 respectively.
In the above-mentioned two sets of proteins, Sox2 and Oct3 are common reprogramming transcription factors. It seems that these two proteins are more important than the other four factors. Actually, iPS cells can be obtained without c-Myc in SOKM factors in both human and mouse fibroblasts.13 However, adding c-Myc led to the recovery of the transformation efficiency of SOK factors.14 With the addition of valproic acid treatment, Sox2 and Oct4 were sufficient to produce iPS cells.15 Other studies showed that c-Myc can be replaced by n-Myc13,16 or by l-myc,13 Sox2 can be replaced by Sox1 or Sox3, whereas Klf4 can be replaced by Klf2 or Klf5.13 Essrb can also take the place of Klf4 and c-Myc in the reprogramming of mouse fibroblasts.17
In addition to the studies on the substitution of reprogramming factors, many other proteins were found to have various impacts on the process of iPS cell generation. Sall4 was identified as being beneficial for the reprogramming.18Wnt3a also increased the efficiency of reprogramming in the absence of c-Myc.19BMP4 was shown to cooperate with Wnt3a to increase the reprogramming of human fibroblast cells.20 Meanwhile, the tumor repressor protein, p53, was found to provide a hurdle that reduces the iPS reprogramming efficiency.21–24
Unfortunately, the efficiency of the reprogramming process is usually very low. Typically only a few iPS cells are generated out of ten thousand or more somatic cells.25 Besides, the mechanisms underlying this transformation are still not very clear. Hence, the search for the crucial interactions and pathways inducing the iPS generation is an important task. Using the traditional structure-based approach for elucidating the molecular mechanisms of the reprogramming factor action has not been very successful, however, mostly due the lack of sufficient information on the 3D structures of these transcription factors.
The main reason for the lack of structural information for these proteins was qualitatively revealed in previous studies, which showed that the majority of transcription factors belong to the class of ![[I with combining low line]](https://www.rsc.org/images/entities/char_0049_0332.gif) ntrinsically
ntrinsically ![[D with combining low line]](https://www.rsc.org/images/entities/char_0044_0332.gif) isordered roteins (IDP), being either disordered as a whole or containing long ntrinsically isordered
isordered roteins (IDP), being either disordered as a whole or containing long ntrinsically isordered ![[R with combining low line]](https://www.rsc.org/images/entities/char_0052_0332.gif) egions (IDR).26–28 By definition, IDPs do not have unique 3D structures in their unbound states under physiological conditions in vitro. Instead, they form dynamic ensembles of mutually transmuting conformations in which the atom positions and backbone Ramachandran angles are not fixed as in ordered proteins, but these positions and angles vary significantly over time with no specific equilibrium values, and the polypeptide chains typically undergo non-cooperative conformational changes over a broad range of time scales.29–32 This highly dynamic nature of IDPs typically precludes them from being crystallized and explains why structural information about transcription factors is rather sparse. Obtaining detailed information on the solution structures of transcription factors, which are often long molecules with complex domain structures, is restricted due to the molecular mass limitations of NMR. Other techniques can only provide low resolution structural information and typically fail at giving structural information at the atomic level. Despite all these difficulties, investigations of IDPs and IDRs are becoming more and more common.33–55
egions (IDR).26–28 By definition, IDPs do not have unique 3D structures in their unbound states under physiological conditions in vitro. Instead, they form dynamic ensembles of mutually transmuting conformations in which the atom positions and backbone Ramachandran angles are not fixed as in ordered proteins, but these positions and angles vary significantly over time with no specific equilibrium values, and the polypeptide chains typically undergo non-cooperative conformational changes over a broad range of time scales.29–32 This highly dynamic nature of IDPs typically precludes them from being crystallized and explains why structural information about transcription factors is rather sparse. Obtaining detailed information on the solution structures of transcription factors, which are often long molecules with complex domain structures, is restricted due to the molecular mass limitations of NMR. Other techniques can only provide low resolution structural information and typically fail at giving structural information at the atomic level. Despite all these difficulties, investigations of IDPs and IDRs are becoming more and more common.33–55
IDPs have been shown to have a number of important biological functions, playing crucial roles in molecular recognition, signaling, and regulation.37,56–61 These functions rely heavily on the conformational flexibility of IDPs.27,29,56,57 More than 150 proteins have been identified as containing functional disordered regions, or being completely disordered, yet performing vital cellular roles.31,56 Twenty-eight separate functions were assigned to these disordered regions, including molecular recognition via binding to other proteins, or to nucleic acids.56,57 An alternative approach was to classify functional disorder into at least five broad groups based on their mode of action.32 Individual studies indicated the roles of intrinsic disorder in many crucial biological processes such as folding and binding,35,62 molecular interaction and recognition,63cell division,54transcription,53 molecular machine activity,44 evolution,64 and human disease.38 Bioinformatics studies on the functional repertoire of IDPs revealed that out of 710 Swiss-Prot functional keywords, 310 keywords were associated with ordered proteins, 238 functional keywords were attributed to disordered proteins, and the remainder 162 keywords yield ambiguity in the likely function-structure associations.59–61 In other studies on the human proteome, disordered regions were found as independent functional units and could be evolutionarily conserved.65 While disordered proteins typically display low sequence conservation,66,67 in contrast some regions of disorder are highly conserved,68–70 probably because such regions are involved in critical functions.66,68,69
Due to their numerous important functional roles in biological systems, IDPs are highly abundant in nature. As indicated by various studies, 7–30% prokaryotic proteins contain long disordered regions of more than 30 consecutive residues, whereas in the eukaryotic proteomes, the number of such proteins reaches 45–50%.31,58,71–78 This observation is in line with the expectation that eukaryotes should have many more proteins involved in signaling and regulation than prokaryotes. Also as a reference, even in the PDB database which is highly biased towards the ordered proteins, around 70% of high resolution protein structures have regions of missing electron density; i.e., contain flexible regions that probably fail to acquire specific structure in the crystals.79 Of which, over 10% have long segments of missing electron density consisting of at least 30 amino acids.80 As of September, 2010, our most recent statistical analysis on over 65![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 PDB proteins determined that 65.7% proteins have at least one missing residue and 7.3% have long disordered regions consisting of at least 30 consecutive residues.
000 PDB proteins determined that 65.7% proteins have at least one missing residue and 7.3% have long disordered regions consisting of at least 30 consecutive residues.
Hence, by taking into consideration the abundance of IDPs and their functional importance, and especially considering the previously elucidated widespread correlation between intrinsic disorder and transcription factors,26–28 it is of interest to evaluate the connection between the intrinsic disorder and the reprogramming transcription factors involved in the generation of iPS from somatic cells. In this paper, we systematically investigated the intrinsic disorder among several sets of proteins that are capable of inducing pluripotent stem cells. As shown by this analysis, all the pluripotent-stem-cell inducing proteins exhibit high amounts of intrinsic disorder, suggesting that there is an intrinsic prerequisite for these transcription factors to be highly flexible and thereby to be able to interact with multiple protein and DNA partners. In addition, the analysis presented here helps to identify the modular nature and functional regions in these factors, thereby providing suggestions for further molecular biology experiments in which the functional subregions are dissected to reveal their specific roles with regard to the conversion from somatic cells into iPS stem cells.
Methods
Selecting the sets of transcription factors
The first study of iPS cells was completed by Takahashi and Yamanaka who used mouse fibroblasts.8 In this study, four transcription factors Oct3/4, Sox2, Klf4, and c-Myc were identified as being critical for iPS generation. In a similar period of time, Yu and Thomson achieved the transformation of human somatic cells into iPS cells by using another four transcription factors: Oct4, Sox2, Nanog, and Lin28.10 Although c-Myc and Lin28 were not present in both sets of transcription factors, the transformation efficiency was tremendously reduced by their removal from their respective sets.10,13,81 Hence, our first dataset includes all of the transcription factors highlighted in the two studies, namely: Sox2, Oct4, c-Myc, Klf4, Lin28, and Nanog.Many other proteins have complex influence on the reprogramming process. For example, Esrrb can replace Klf4 and c-Myc;17 Sox1 or Sox3 are able to substitute for Sox2; Either Klf 2 or Klf5 can take the place of Klf4; l-Myc or n-Myc can supplant c-Myc.13,16 In addition, it is worth noting that increased p53 levels reduce the iPS reprogramming efficiency.21–24 In contrast, Sall4, Wnt3a, and BMP4 are beneficial for the process.18,19 Therefore, all these effector proteins are grouped into a second dataset.
The third dataset includes a collection of ES cell-related factors, also called ES markers, that were found to be over-expressed in embryonic stem cells by Takahashi and Yamanaka.9 The original collection had 24 factors. Following further study, four of these became the SOKM group.12 Nanog, which became part of the SOLN group,12 and the reprogramming effector Sall4 were also in this set of 24 factors. After removing these 6 factors, the remaining 18 are included in the third dataset, namely: Ecat1; Dppa5 (Esg1); Fbxo15; Eras; Dnmt3l; Ecat8; Gdf3; Sox15; Dppa4; Dppa2; Fthl17; Rex1 (Zpf42); Utf1; Tcl1; Dppa3 (Stella); β-catenin; Stat3; and Grb2.
Due to the fact that most reprogramming experiments were performed in mouse models, protein sequences from Mus musculus are used throughout this study. All these sequences and their SwissProt IDs are listed in Table 1. Also included in Table 1 are the summary disorder predictions as discussed below.
| Sequence | Structure | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Protein Name | SwissProt ID | L | IDAA (%) | Q | PDB ID | Seq. Iden. | Position | Frac. of Struct. | Method |
| The sequence- and structure-related information was extracted from SwissProt and PDB databases. In the cases when multiple PDB structures were available for the same SwissProt sequence, the PDB structure with the longest segment and highest resolution and earliest experimental date was used. * Indicates a template structure in Swiss-Model Repository. When the sequences identity is over 40%, all the templates are listed. Otherwise, only the template with the highest sequence identity was filled in the table. IDAA % corresponds to the percentage of amino acid residues predicted to be disordered by PONDR® VSL2. Q1, Q2, Q3, and Q4 indicates the quadrant number in CH–CDF plot for each protein: proteins in Q1 are structured by CDF, but disordered by CH; proteins in Q2 are predicted to be structured by both CDF and CH; proteins in Q3 are disordered by CDF but structured by CH; and Q4 is for disordered proteins predicted by both methods. “Fraction of structure” means the percentage of amino acids with known structures. | |||||||||
| Sox2 | P48432 | 319 | 100% | 4 | 1GT0 | — | 41–120 | 25.1% | X-ray |
| Oct4 | P20263 | 352 | 67.3% | 3 | 1POU* | 69% | 137–205 | 19.6% | NMR |
| 1OCP | — | 217–282 | 18.8% | NMR | |||||
| Klf4 | Q60793 | 483 | 96.7% | 3 | 2WBS | — | 395–483 | 18.4% | X-ray |
| c-Myc | P01108 | 439 | 79.5% | 4 | 1NKP* | 92% | 353–434 | 18.7% | X-ray |
| Lin28 | Q8K3Y3 | 209 | 76.6% | 3 | 1H95* | 40% | 37–113 | 36.8% | NMR |
| 2CQF* | 94% | 136–186 | 24.4% | NMR | |||||
| Nanog | Q80Z64 | 305 | 62.6% | 3 | 2VI6 | — | 96–155 | 19.7% | X-ray |
| Esrrb | Q61539 | 433 | 37.2% | 2 | 1LO1* | 97% | 97–186 | 20.8% | NMR |
| 2GPO* | 74% | 211–433 | 51.5% | X-ray | |||||
| Sox1 | P53783 | 391 | 94.9% | 3 | 1GT0* | 96% | 49–127 | 20.2% | X-ray |
| Sox3 | P53784 | 375 | 87.2% | 3 | 1GT0* | 91% | 67–145 | 21.1% | X-ray |
| Klf2 | Q60843 | 354 | 92.7% | 3 | 2WBS* | 91% | 269–353 | 24.0% | X-ray |
| Klf5 | Q9Z0Z7 | 446 | 77.1% | 3 | 2EBT* | 94% | 352–446 | 21.3% | NMR |
| l-Myc | P10166 | 368 | 74.7% | 4 | 1NKP* | 47% | 285–368 | 22.8% | X-ray |
| n-Myc | P03966 | 462 | 80.5% | 3 | 1NKP* | 54% | 379–462 | 18.2% | X-ray |
| p53 | P02340 | 387 | 64.6% | 3 | 2IOI | — | 92–289 | 51.2% | X-ray |
| 1AIE* | 90% | 320–349 | 7.8% | X-ray | |||||
| Sall4 | Q8BX22 | 1067 | 71.5% | 3 | 2ELS* | 57% | 388–413 | 2.4% | NMR |
| 2EOS* | 41% | 405–445 | 3.8% | NMR | |||||
| 2JP9* | 41% | 572–655 | 7.9% | NMR | |||||
| 1SP2* | 51% | 878–906 | 2.7% | NMR | |||||
| 2EPU* | 48% | 900–930 | 2.9% | NMR | |||||
| Wnt3a | P27467 | 352 | 31.8% | 2 | — | — | — | — | — |
| BMP4 | P21275 | 408 | 46.8% | 3 | 2H62* | 91% | 307–408 | 25.0% | X-ray |
| Ecat1 | Q9CWU5 | 440 | 66.1% | 3 | — | — | — | — | — |
| Dppa5 | Q9CQS7 | 118 | 28.8% | 2 | — | — | — | — | — |
| Fbxo15 | Q9QZN0 | 433 | 14.3% | 2 | 2AST* | 33% | 1–42 | 9.7% | X-ray |
| Eras | Q7TN89 | 227 | 29.5% | 2 | 6Q21* | 47% | 39–201 | 71.8% | X-ray |
| Dnmt3l | Q9CWR8 | 421 | 29.9% | 2 | 2PV0* | 66% | 68–415 | 82.7% | X-ray |
| Ecat8 | Q9CWU0 | 1215 | 31.1% | 2 | 1EJF* | 24% | 1075–1177 | 8.5% | X-ray |
| Gdf3 | Q07104 | 366 | 11.7% | 2 | 3BMP* | 51% | 263–366 | 28.4% | X-ray |
| Sox15 | P43267 | 231 | 100% | 4 | 1O4X* | 77% | 45–121 | 33.3% | NMR |
| Dppa4 | Q8CCG4 | 296 | 58.8% | 3 | — | — | — | — | — |
| Dppa2 | Q9CWH0 | 301 | 62.5% | 3 | — | — | — | — | — |
| Fthl17 | Q99MX2 | 176 | 11.9% | 2 | 2CIH* | 54% | 5–176 | 97.7% | X-ray |
| Rex1 | P22227 | 288 | 77.1% | 3 | 1UBD* | 75% | 170–282 | 39.2% | X-ray |
| Utf1 | Q6J1H4 | 339 | 86.1% | 3 | — | — | — | — | — |
| Tcl1 | P56280 | 116 | 17.2% | 2 | 1JNP | — | 1–116 | 100% | X-ray |
| Dppa3 | Q8QZY3 | 150 | 87.3% | 4 | — | — | — | — | — |
| β-catenin | Q02248 | 781 | 30.0% | 2 | 2G57* | 100% | 19–44 | 3.3% | NMR |
| 1L7W | — | 134–671 | 68.9% | X-ray | |||||
| Stat3 | P42227 | 770 | 32.3% | 2 | 1BG1 | — | 127–715 | 76.5% | X-ray |
| 1YVL* | 52% | 2–689 | 89.4% | X-ray | |||||
| Grb2 | Q60631 | 217 | 20.3% | 2 | 1GBQ | — | 1–59 | 27.2% | X-ray |
| 1JYR* | 100% | 60–151 | 42.4% | X-ray | |||||
| 2VVK* | 100% | 161–214 | 24.9% | X-ray | |||||
Disorder prediction
The intrinsic disorder propensities of the analyzed proteins were evaluated by two different disorder predictors. The first one is PONDR® VL-XT,73 which applies various compositional probabilities and hydrophobic measures of amino acid as the input features of artificial neural networks for the prediction. Although it is no longer the most accurate predictor, it is very sensitive to the local compositional biases. Hence, it is capable of identifying potential molecular interaction motifs.75,82 The second predictor is a meta-predictor PONDR®FIT,77 that combines six individual predictors, which are PONDR® VL-XT,73VSL2,83VL3,84 FoldIndex,85 IUPred,86 and TopIDP.87 This meta-predictor is moderately more accurate than each of the component predictors.Molecular recognition feature (MoRF)
Being defined as a short order-prone motif within a long disordered region and being able to undergo disorder-to-order transition during the binding to a specific partner,![[M with combining low line]](https://www.rsc.org/images/entities/char_004d_0332.gif)
![[o with combining low line]](https://www.rsc.org/images/entities/char_006f_0332.gif) lecular ecognition
lecular ecognition ![[F with combining low line]](https://www.rsc.org/images/entities/char_0046_0332.gif) eature (MoRF) usually has much higher content of aliphatic and aromatic amino acids than disordered regions in general. Due to these peculiarities, MoRF regions are frequently observed as sharp dips in the corresponding plots representing per-residue distribution of PONDR® VL-XT disorder scores. Hence, based on the PONDR® VL-XT prediction and a number of other attributes, the MoRF regions can be identified.75,82
eature (MoRF) usually has much higher content of aliphatic and aromatic amino acids than disordered regions in general. Due to these peculiarities, MoRF regions are frequently observed as sharp dips in the corresponding plots representing per-residue distribution of PONDR® VL-XT disorder scores. Hence, based on the PONDR® VL-XT prediction and a number of other attributes, the MoRF regions can be identified.75,82
ANCHOR analysis
In addition to MoRF identifiers, potential binding sites in disordered regions can be identified by the ANCHOR algorithm.88,89 This approach relies on the pairwise energy estimation approach developed for the general disorder prediction method IUPred,86,90 and is based on the hypothesis that long regions of disorder contain localized potential binding sites that cannot form enough favorable intrachain interactions to fold on their own, but are likely to gain stabilizing energy by interacting with a globular protein partner.88,89 Here we are using the term ANCHOR-indicated binding site (AiBS) to identify a region of a protein suggested by the ANCHOR algorithm to have significant potential to be a binding site for an appropriate but typically unidentified partner protein.CH–CDF plot
CH–CDF plot is an analytical tool combining outputs of two binary predictors,harge-![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) ydropathy (CH) plot30,74 and umulative istribution unction (CDF) plot, both predicting an entire protein as being ordered or disordered.71,74 The CH-plot places each protein onto a 2D graph as a single point by taking the mean Kyte-Doolittle hydropathy91 of a protein as its X-coordinate and the mean net charge of the same protein as its Y-coordinate. In a CH-plot, structured, globular proteins and fully disordered, highly polar proteins were shown to be separated by a boundary line. Of the training set examples, the proteins located above this boundary are likely to be disordered, while proteins located below this line are likely to be structured. The vertical distance on CH-plot from the location of the protein to the boundary line is then a scale of disorder (or structure) tendency of the protein. Here this distance is referred to as the CH-distance.
ydropathy (CH) plot30,74 and umulative istribution unction (CDF) plot, both predicting an entire protein as being ordered or disordered.71,74 The CH-plot places each protein onto a 2D graph as a single point by taking the mean Kyte-Doolittle hydropathy91 of a protein as its X-coordinate and the mean net charge of the same protein as its Y-coordinate. In a CH-plot, structured, globular proteins and fully disordered, highly polar proteins were shown to be separated by a boundary line. Of the training set examples, the proteins located above this boundary are likely to be disordered, while proteins located below this line are likely to be structured. The vertical distance on CH-plot from the location of the protein to the boundary line is then a scale of disorder (or structure) tendency of the protein. Here this distance is referred to as the CH-distance.
CDF is also a binary tool to describe the disorder status of an entire protein.71,74 In brief, it is a cumulated histogram of residues at various disorder scores. By definition, an ordered protein has more order-promoting residues and less disorder-promoting residues. Therefore, the CDF curve of a structured protein would increase very quickly in the domain of low disorder scores, and then goes flat in the domain of high disorder scores. For disordered proteins, the CDF curve would go upward slightly in the domain of low disorder scores, then increase quickly in the domain of high disorder scores. Hence, on the 2D CDF-plot, ordered protein curves tend to stay on the upper left half, whereas disordered protein curves tend to locate at the lower right half of the plot. By comparing the locations of CDF curves for a group of fully disordered and fully ordered proteins, a boundary line between these two groups of proteins can be identified. This boundary then could be used for evaluating the disorder status of a given protein. In fact, proteins whose CDF curves are located above the boundary line are likely to be structured, while proteins with CDF curves below the boundary are likely to be disordered. The averaged distance from the CDF curve to this boundary is also a measure of the disorder (order) status of a given protein and is referred to as CDF-distance. CDF-plots based on various disorder predictors have different accuracies.92 In our previous study, PONDR® VSL2-based CDF achieved the highest accuracy which was up to 5–10% higher than the second best of the other five CDF functions for the separation of fully disordered proteins from structured proteins also containing disordered loops or tails. As for the separation of fully structured from fully disordered proteins, the CDF curves derived from the various disorder predictors all exhibited similar accuracies.92
By putting together both the CH-distance and the CDF-distance, a new method called the CH–CDF plot was developed.93 The CH–CDF plot provides very useful information on the general disorder status of a given protein. After setting up boundaries at CH = 0 and CDF = 0, the entire CH–CDF plot can be split into four quadrants. Starting from the upper right quadrant, by taking the clockwise sequence, the four quadrants are named Q1 (upper right), Q2 (lower right), Q3 (lower left), and Q4 (upper left). Proteins in Q1 are structured by CDF, but disordered by CH; proteins in Q2 are predicted to be structured by both CDF and CH; proteins in Q3 are disordered by CDF but structured by CH; and proteins in Q4 are predicted to be disordered by both methods. The location of a given protein in this CH–CDF plot gives information about its overall physical and structural characteristics as discussed below.
Results
The 3D structural information for three protein datasets
Table 1 and Fig. 1 present the basic sequence and structure information for all the proteins in three datasets and shows that out of 35 proteins only 9 have PDB structures, of which only one is for the entire sequence and the other 8 are for protein fragments: three structures are for protein segments longer than 50% of the original sequences, and the last five structures cover just ∼20% of the original sequences. Hence, the experimental information on the 3D structure of these proteins is very limited. Through sequence alignment, another 19 proteins can be matched to proteins with known structures. However, in homologue structures, the sequence coverage is normally at the level of about 20%, and only five proteins have sequence coverage higher than 50%. The templates for these five proteins have sequence identity with original protein sequences of 50% or higher. Hence, these five structures are more reliable than others. By taking into account of all above cases, the total number of structure-known proteins with sequence coverage higher than 50% is also nine. As a result, although the information for partially known structures increased after taking into account of both experimental and computational structures, the available structural information for entire sequence is still quite limited.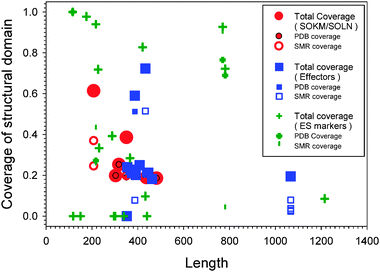 | ||
| Fig. 1 Coverage of structured domains in three sets: SOKM/SOLN (red symbols), Effectors (blue symbols), and ES markers (green symbols). The coverage is defined as the ratio between length of structured region(s) and full length of the sequence. Each protein may have corresponding PDB structures and Swiss-Model Repository (SMR) homologous structures. Therefore, there are symbols corresponding to the PDB and SMR coverage, respectively. The total coverage is the sum of PDB and SMR coverage. | ||
Fig. 1 provides a summary of the structural coverage for proteins with known 3D structures. Here, the percentage of each sequence that aligned with one or more 3D structures in PDB (PDB coverage) and/or in Swiss-Model Repository of homologous structures (SMR coverage) is plotted versus the length of each protein. The plot includes data for SOKM/SOLN (red symbols), Effectors (blue symbols), and ES markers (green symbols). A decreased structural coverage of the SOKM/SOLN and effector proteins compared to the structural coverage of the ES markers is evident. One of the major reasons for the lack of 3D structures likely comes from the intrinsically disordered nature of these proteins. Therefore, intrinsic disorder-based analysis has to be applied to fully understand structures and functions of these proteins.
According to their SwissProt annotations, several proteins in the analyzed datasets belong to five protein families: Sox (Sox1, Sox2, Sox3, Sox15); Myc (c-Myc, l-Myc, n-Myc); Klf (Klf2, Klf4, Klf5); Dppa (Dppa2, Dppa3, Dppa4, Dppa5); and Ecat (Ecat1, Ecat2, Ecat3, Ecat4, Ecat5, Ecat8). As indicated by the results of the reprogramming studies, proteins in the same family can be simultaneously over-expressed in the reprogramming process,8,94,95 as well as be replaced by other proteins in the same family.10,13,81
CH–CDF plot
The CH–CDF plot for the three sets of proteins studied herein is shown in Fig. 2(a), with the separate CH and CDF plots in Fig. 2(b) and (c), respectively. The three sets are: six iPS-inducing factors (SOKM and SOLN); eleven effectors influencing the reprogramming efficiency; and eighteen ES cell markers. Apparently, ES markers and effectors are located mostly in Q2 and Q3, respectively. Proteins in Q2 are predicted to be structured by both CH and CDF analyses. These proteins are structured and compact. Proteins in Q3 are predicted to be disordered by CDF, but compact by CH-plot. One possibility that should be tested is that such a protein might be a native molten globule. A second possibility is that such proteins contain mixtures of structured and disordered regions, a possibility that is supported for many of these proteins by the data in Table 1 and Fig. 1. Proteins in Q4 are likely to be mostly disordered and extended if their charge is sufficiently high. Each dataset has one or two proteins in this category. This is an indication that proteins related to various aspects of reprogramming are somehow connected to intrinsic disorder.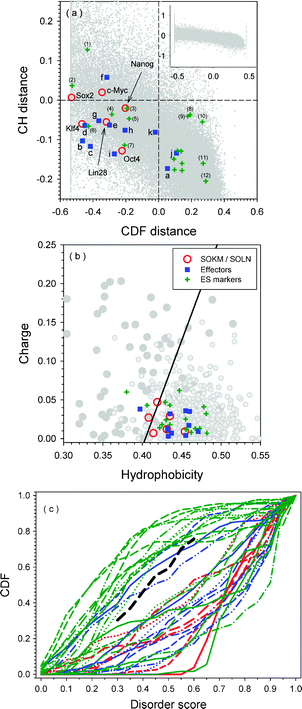 | ||
| Fig. 2 Panel (a): CH–CDF plot for proteins in three datasets. The CDF distance is calculated by PONDR-VSL2 CDF function. CH distance is the normal distance on Charge-Hydropathy plot. Red circles are SOKM/SOLN proteins as indicated by their names in the plot. Blue squares are various effectors: (a) Essrb; (b) Sox1; (c) Sox3; (d) Klf2; (e) Klf5; (f) l-myc; (g) n-Myc; (h) P53; (i) Sall4; (j) Wnt3a; and (k) BMP4. Green pluses are 18 different markers. However, only 12 are labeled by numbers: (1) Dppa3; (2) Sox15; (3) Dppa2; (4) Rex1; (5) Dppa4; (6) Utf1; (7) Ecat1; (8) Grb2; (9) Tcl1; (10) Fthl17; (11) Gdf3; and (12) Fbxol5. Other 6 proteins are all located close to each other in the second quadrant. By their values on CH distance from big to small, they are: Stat3; Dnmt3l; Dppa5; Ecat8; Eras; and β-Catenin. Panel (b): Charge-Hydropathy plot for three sets of proteins. The dashed line is the boundary between intrinsically disordered proteins and structured proteins. Proteins above the boundary line are disordered, while proteins below the boundary are structured. The trapezium above the boundary line surrounded by dotted lines is very frequently occupied by disordered proteins, while the trapezium below the boundary is often taken by structured proteins. Panel (c): PONDR® VSL2-based CDF plot analysis of proteins from three sets: SOKM/SOLN (red lines), Effectors (blue lines), and ES markers (green lines). The dashed black line is the boundary between fully disordered and fully structured proteins. | ||
All the reprogramming transcription factors have negative CDF-distances (Fig. 2(a) and (c)); i.e., they are predicted to be intrinsically disordered by the PONDR® VSL2-based CDF predictor. As a comparison, 8 of 11 effectors and 7 of 18 ES makers have negative CDF-distances. These results suggest that the reprogramming function requires structural flexibility and therefore relies on intrinsic disorder.
The evaluation from the CH-plots in Fig. 2(a) and (b) gives slightly different results compared to those from the CDF-plots in Fig. 2(a) and (c). The CH-distances for Sox2 and c-Myc are greater than zero, indicating complete disorder. The other factors have CH distances below zero, indicating structure, but most are close to the boundary. That is, of the six reprogramming factors, only Oct4 has a CH-distance below −0.1. The other three proteins are in the range between 0 and −0.1. In the set of effectors, only l-Myc has the CH-distance above zero, whereas five out of eleven are below −0.1. The rest of the five effectors are in the middle region from 0 and −0.1. Finally, half of the proteins in the group of ES markers have CH-distances lower than those of reprogramming factors and effectors.
Disorder prediction and functional regions of iPS inducing factors
Fig. 3 represents the residue-based disorder predictions for both sets of iPS-inducing transcription factors: SOKM and SOLN. In these plots, all residues/regions with a disorder score higher than 0.5 are predicted to be disordered. By taking into consideration the fact that a structural domain normally requires approximately 50 or more consecutively residues to be ordered, it can be observed that all these proteins are highly disordered. Only the segments of Oct4 (residues 140–200), Lin28 (residues 30–120), and Nanog (residues 100–150) contain possible structured domains (see Fig. 3(b), (e), and (f)). Actually, this conclusion is in agreement with the available structural data which show that these three regions have PDB structures as shown in Table 1 and Fig. 4. The region 137–205 of Oct4 has sequence identity of 69% with the structured POU domain of Oct1 (PDBid: 1POU). Segment 37–113 of Lin28 has 40% sequence identity with the structured fragment of the YB-1 protein (PDBid: 1H95). Also, the homeodomain of Nanog (residues 96–155) also has crystal structure (PDBid: 2VI6). Hence, all the predictions on structured domains are verified as correct by experiments.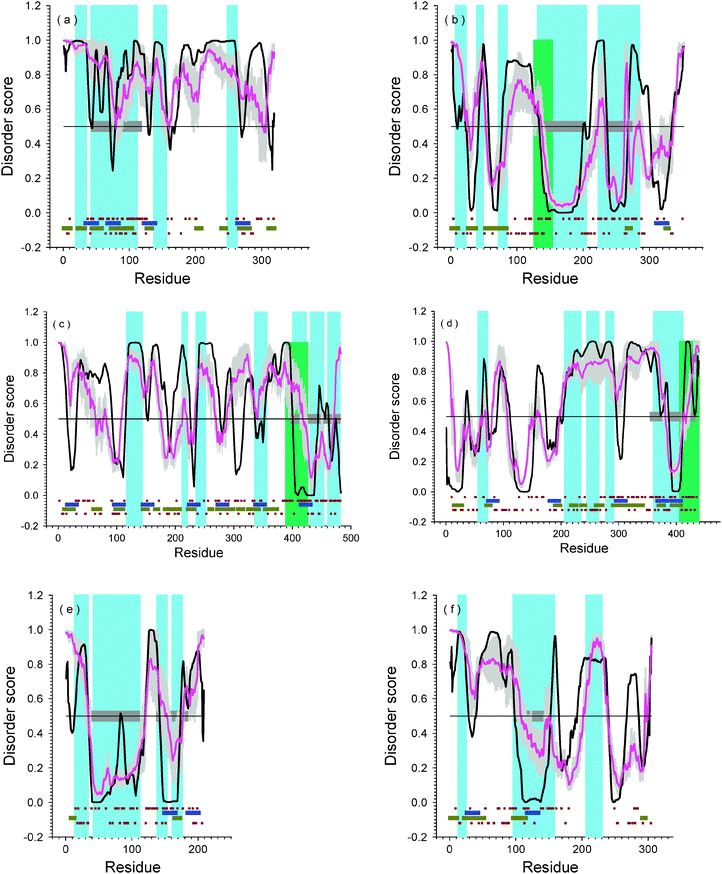 | ||
Fig. 3 Disorder predictions for iPS-inducing factors: (a) Sox2; (b) Oct4; (c) Klf4; (d) c-Myc; (e) Lin28; (f) Nanog. The black lines are disorder prediction from PONDR®VL-XT; pink lines are prediction of PONDR-FIT; the grey shadows are the prediction error of PONDR-FIT. Light blue and light green regions are SMART domains with high confidence or low threshold values. Horizontal blue bars are potential MoRF regions predicted by MoRF-II predictor.82 Horizontal dark yellow bars are binding motifs identified by ANCHOR.83,84 Brown dots nearby blue bars are locations of positively charged residues (up) and negatively charged residues (down). The horizontal gray bars are domains with known PDB structures. The SMART regions are: (a) AA19-AA35: ![[L with combining low line]](https://www.rsc.org/images/entities/char_004c_0332.gif) ow omplexity egion (LCR); AA42–AA112: HMG; AA137–AA156: LCR; AA248–AA263: LCR. (b) AA7–AA23: LCR; AA39–AA49: LCR; AA72–AA85: LCR; AA126–AA153: coiled coil; AA131–AA205: POU domain; AA223–AA285: HOX domain. (c) AA116–AA144: LCR; AA211–AA221: LCR; AA235–AA252: LCR; AA335–AA357: LCR; AA388–AA427: Zinc ginger BED domain; AA400–AA424: Zinc finger; AA430–AA454: Zinc Finger; AA460–AA482: Zinc finger. (d) AA55–AA73: LCR; AA203–AA237: LCR; AA242–AA268: LCR; AA275–AA294: LCR; AA360–AA412: bHLH; AA406–439: coiled coil. (e) AA14–AA35: LCR; AA41–AA112: CSP; AA138–AA154: Zinc finger; AA160–AA176: Zinc finger. (f) AA13–25: LCR; AA96–158: HOX; AA206–230: LCR. ow omplexity egion (LCR); AA42–AA112: HMG; AA137–AA156: LCR; AA248–AA263: LCR. (b) AA7–AA23: LCR; AA39–AA49: LCR; AA72–AA85: LCR; AA126–AA153: coiled coil; AA131–AA205: POU domain; AA223–AA285: HOX domain. (c) AA116–AA144: LCR; AA211–AA221: LCR; AA235–AA252: LCR; AA335–AA357: LCR; AA388–AA427: Zinc ginger BED domain; AA400–AA424: Zinc finger; AA430–AA454: Zinc Finger; AA460–AA482: Zinc finger. (d) AA55–AA73: LCR; AA203–AA237: LCR; AA242–AA268: LCR; AA275–AA294: LCR; AA360–AA412: bHLH; AA406–439: coiled coil. (e) AA14–AA35: LCR; AA41–AA112: CSP; AA138–AA154: Zinc finger; AA160–AA176: Zinc finger. (f) AA13–25: LCR; AA96–158: HOX; AA206–230: LCR. | ||
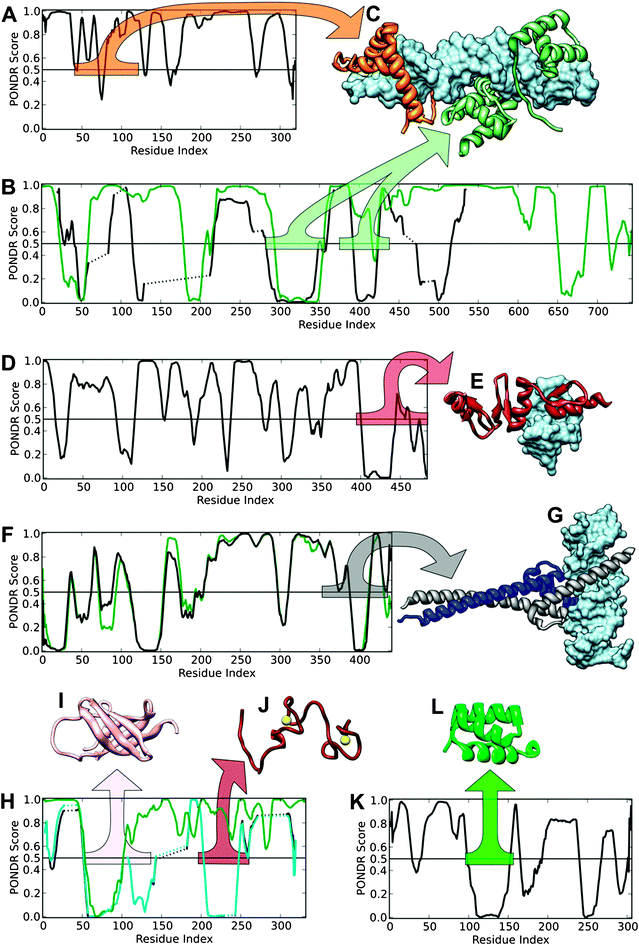 | ||
| Fig. 4 PONDR®VL-XT predictions and structures of mouse reprogramming transcription factors and their homologues. VLXT predictions for mouse proteins (black lines) are shown, along with predictions for the aligned human homologues (green lines) for Oct4 (B), c-Myc (F) and Lin28 (H). Structures depict domains from mouse reprogramming proteins and their homologues as variously colored ribbons, with a corresponding colored bar in the disorder prediction plot indicating the source region. In complexes with DNA, DNA is depicted as a blue surface. Disorder predictions for Sox2 (A) and oct4 and its human homolog (P14859) are shown (B) with the complex between Sox2, the human homolog of mouse Oct4, and DNA (C, PDB ID1GT0). Disorder prediction for Klf4 is shown (D) with its complex with DNA (E, PDB ID2WBS). Disorder prediction for c-Myc and its human homolog (P01106) is show (F) with its complex with max and DNA (G, PDB ID1NKP). Disorder predictions for Lin28, its human homolog (Q9H9Z2), and similar protein Y-box-binding protein (YBX, P67809, cyan line) is show (H) with the structures of the cold shock domain of YBX (I, PDB ID1H95) and the zinc-finger domain of human Lin28 (J, PDB ID2CQF). Disorder prediction for Nanog is shown (K) with the structure of its homeodomain domain (L, PDB ID2VI6). | ||
On the other hand, Table 1 and Fig. 4 show several additional structured domains other than the above mentioned three structures. Nonetheless, these additional structures come from complexes or correspond to the shorter structured regions in the prediction. For example, the 41–120 region of Sox2 has a PDB structure that corresponds to a complex of three different molecules (PDBid: 1GT0). The solution structure of the 217–282 fragment of Oct4 was obtained by NMR as an ensemble of 20 conformations (PDBid: 1OCP). All these conformations are rather different from each other in terms of absolute spatial coordinates. Besides, the Oct4 240–270 fragment is predicted to be structured in Fig. 3(b), although this region has only 30 residues. Similarly to Oct4, there is a significant correlation between order/disorder prediction and actual structural data for Klf4: the 400–440 fragment of Klf4 is predicted to be structured, and the 395–483 region of Klf4 forms a zinc-finger that has an X-ray structure (PDBid: 2WBS) in a complex with DNA molecules. The c-Myc fragment containing residues 390–410 is also predicted to be structured. This region was crystallized as a complex with Max and DNA (PDBid: 1NKP). In Lin28, the 136–186 fragment has 94% sequence identity to its human homologue, for which a solution structure was resolved by NMR (PDBid: 2CQF). In the ensemble of solution structures, both termini of this region are highly flexible. In the prediction, the 145–155 region of mouse Lin28 is predicted to be structured. Hence, essentially all of the structural information in Table 1 shows excellent correspondence with the regions or domains predicted to be ordered by the disordered protein analysis (see Fig. 4).
Fig. 3 shows that both Oct4 and c-Myc have short regions of predicted order without any structural annotation. These are fragment 300–340 of Oct4, and segments 1–30 and 110–150 of c-Myc. However, the functions and actual structures of these regions are still unknown. We propose that these segments would be good candidates for structure-determination efforts. In addition, by taking into consideration of the lengths, which are shorter than typical structured domains, these segments would also be candidates to form structure in complexes with other proteins or DNA.
Fig. 3 also shows that all six proteins contain a large number of potential disorder-based binding sites, α-MoRFs and ANCHOR-indicated binding sites (AiBSs), which often completely or partially overlap with each other. The fact that these factors possess numerous binding sites clearly indicates that their major function is to be involved in interaction with various binding partners.
Difference in disorder prediction for proteins in the same family
Fig. 5 represents the comparison of disorder predictions for the members of four protein families: Sox, Myc, Klf, and Dppa. The Ecat family was excluded from this analysis due to the low sequential and functional similarities of its members. The analysis based on the PONDR®VL-XT disorder prediction is very informative since the prediction curve characterizes not only the general conformational properties of the protein chains and its disorder propensity, but also the possible interaction regions, MoRFs and AiBSs. Fig. 5 shows that within each family MoRFs and AiBSs often overlap. Since there is typically a good agreement between MoRF and AiBSs predictions, in most cases the two features will be considered together rather than separately.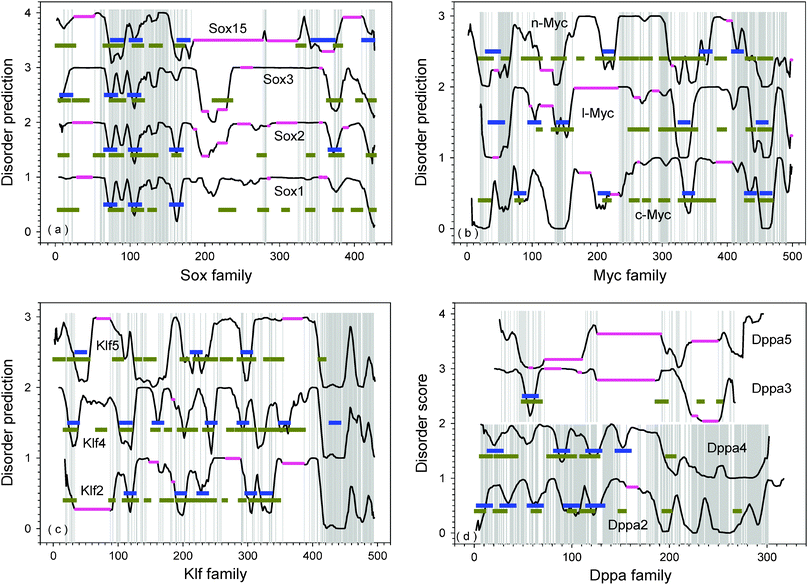 | ||
| Fig. 5 Comparison of disorder prediction for proteins in the same family: (a) Sox; (b) Myc; (c) Klf; and (d) Dppa. The pink horizontal lines are alignment indels. Horizontal blue bars are potential MoRF regions predicted by MoRF-II predictor.82 Horizontal dark yellow bars are binding motifs identified by ANCHOR.83,84 The disorder prediction was conducted by PONDR®VL-XT. Normally, residues with score higher than 0.5 are disordered, while residues with score lower than 0.5 are structured. However, the disorder scores on y-axis were shifted upwards to separate various proteins. The corresponding intervals on y-axis are labeled behind the name of each protein. (a) For Sox family: Sox1 (0–1); Sox2(1–2); Sox3(2–3); and Sox15(3–4). (b) For Myc family: c-Myc(0–1); l-Myc(1–2); and n-Myc(2–3). (c) For Klf family: Klf2(0–1); Klf4(1–2); and Klf5(2–3). (d) For Dppa family: Dppa2(0–1); Dppa4(1–2); Dppa3(2–3); and Dppa5(3–4). The threshold between disordered and structured residues should also be shifted at the medium of each interval, accordingly. The details of the aligned sequences are shown in the ESI, Fig. S2.‡ | ||
RY-related HMG b![[x with combining low line]](https://www.rsc.org/images/entities/char_0078_0332.gif) . SRY is the abbreviation of ex-determining egion
. SRY is the abbreviation of ex-determining egion ![[Y with combining low line]](https://www.rsc.org/images/entities/char_0059_0332.gif) , which is a sex-determining gene on Y chromosome in both marsupial and placental mammals.96 The product of this gene is a transcription factor responsible for the initiation of male sex determination. This protein belongs to the igh obility roup (HMG) family, members of which contain HMG-box domain(s) that bind to DNA and which change the conformation of chromatin.97 The position of this domain in the Sox2 sequence is from residue 40 to residue 120. The global fold of an HMG-box domain is an ‘L-shaped’ structure with three α-helices, the long arm comprising the N-terminal extended strand and helix III, and the short arm comprising helices I and II.97Proteins containing the HMG-box can bind to non-B-type DNA conformations only and with high affinity.98 Sox proteins were also reported to bind DNA sequence containing the WWCAAW (W = A/T) motif and similar sequences with low affinity.99 The structure of the HMG domain of Sox2 was determined as part of a ternary complex with the POU domain of Oct4 and DNA (PDB id: 1GT0).
, which is a sex-determining gene on Y chromosome in both marsupial and placental mammals.96 The product of this gene is a transcription factor responsible for the initiation of male sex determination. This protein belongs to the igh obility roup (HMG) family, members of which contain HMG-box domain(s) that bind to DNA and which change the conformation of chromatin.97 The position of this domain in the Sox2 sequence is from residue 40 to residue 120. The global fold of an HMG-box domain is an ‘L-shaped’ structure with three α-helices, the long arm comprising the N-terminal extended strand and helix III, and the short arm comprising helices I and II.97Proteins containing the HMG-box can bind to non-B-type DNA conformations only and with high affinity.98 Sox proteins were also reported to bind DNA sequence containing the WWCAAW (W = A/T) motif and similar sequences with low affinity.99 The structure of the HMG domain of Sox2 was determined as part of a ternary complex with the POU domain of Oct4 and DNA (PDB id: 1GT0).
By comparing the disorder predictions after sequence alignment in Fig. 5(a), it can be seen that the PONDR plots of Sox1, Sox2, and Sox3 are highly similar to each other. The N-terminal region contains a highly conserved HMG-box domain (residues 70–150). This domain is highly conserved among Sox1, Sox2, Sox3, and Sox15 proteins. From structural point of view, the HMG-box domain is composed of three helices separated by short loops. The first two helices are actually predicted to be MoRFs (and/or AiBSs) as shown in Fig. 5(a). Following this domain, there is another predicted MoRF/AiBS in Sox2 (around residue 160). This potential binding motif was also predicted in Sox1 and Sox15, but not Sox3. This corresponding dip in the PONDR® VL-XT curve is deep in Sox1; much shallower in Sox3; and is twisted into structure-prone regions in Sox15. There are another two dips in the regions 190–230 and 370–380. These dips are shallower in Sox1, and deeper in Sox3. The corresponding region in Sox15 was absent or shifted towards the N-terminal part of the molecule. The second dip was also a predicted MoRF/AiBS in Sox2. Just before this second dip is another similar segment among the Sox proteins at residues 340–355. This region is predicted to be a MoRF/AiBS in Sox15. Sox proteins also have a conserved hydrophobic motif VPLTHI at the very end of their C-termini which forms a dip and may also serve as a potential interaction site. Proline and histidine in this motif are conserved throughout the family. Three aliphatic residues can be replaced by other similar residues. Threonine was mostly conserved but substituted by Serine in Sox2. This conserved region was predicted to be a MoRF in Sox15.
Compared to Sox2, both Sox1 and Sox3 have several poly-Ala insertions and/or Gly-rich regions at various locations. As indicated by our short peptides predictor SPA,76 both poly-Ala and Gly-rich regions have moderately higher tendencies of being disordered as compared to segments from structured proteins. By taking into consideration that these poly-Ala and Gly-rich regions are just before or after MoRFs/AiBSs, the binding of these regions to their partners will definitely be affected. First, the poly-Ala and Gly-rich regions would tend to increase the exposure or accessibility of the nearby MoRFs/AiBSs. Second, the regions that flank MoRFs/AiBSs appear in many cases to increase binding affinity by weak interactions,100 but poly-Ala and Gly-rich regions seem unlikely to make such weak interactions, so MoRFs/AiBSs with such flanking regions might, overall, bind more weakly as compared to similar binding regions set in other contexts.
Compared to other Sox proteins, Sox15 has several long deletions at its C-terminal region. Two of the deletions cover a region corresponding to the last MoRF of Sox2. Right after this region, Sox15 has another predicted MoRF at the very C-terminal end. Hence, the C-terminal region of Sox15 is shorter than in other members of the Sox family, but it has two closely spaced MoRFs. This sequential structural feature makes Sox 15 apparently different from other Sox family proteins. Actually, Sox1, Sox2, and Sox3 belong to SoxB1 subgroup, while Sox15 is the only member of the subgroup SoxG.101
![[b with combining low line]](https://www.rsc.org/images/entities/char_0062_0332.gif) asic elix-oop-elix eucine
asic elix-oop-elix eucine ![[Z with combining low line]](https://www.rsc.org/images/entities/char_005a_0332.gif) ipper) domain.105 In c-Myc, the position of the basic motif is in the vicinity of residues 350–370, followed by the HLH motif in the 370–410 region, and the Leucine Zipper in the 410–430 region.
ipper) domain.105 In c-Myc, the position of the basic motif is in the vicinity of residues 350–370, followed by the HLH motif in the 370–410 region, and the Leucine Zipper in the 410–430 region.
For Myc family proteins in Fig. 5(b), the most conserved pattern of disorder prediction is seen at the C-terminal region. Residues 410–490 represent the highly conserved bHLH leucine zipper domain. This conserved region is rich of predicted MoRFs and AiBSs. c-Myc has two predicted MoRFs in this region, while l-Myc and n-Myc have one each. The Leuzine Zipper region normally forms homo- or hetero-dimers to interact with DNA.106 All three Myc proteins have another MoRF in the upstream region not far away from the bHLH/LZ domain. This MoRF in l-Myc is separated from bHLH/LZ by a poly-Glu segment. The poly-Glu segment is predicted to be highly disordered by SPA.76 Further upstream of this MoRF, these three proteins all have a conserved acidic region. Although highly conserved, this acidic-rich region in c-Myc and n-Myc is much longer than in l-Myc.
Besides above-mentioned conserved region at C-terminal, there are three other major conserved regions across the Myc proteins: 50–70; 135–150; and 295–310. Although these conserved regions correspond to various dips, only l-Myc has a predicted MoRF in the 135–150 region. l-Myc contains three other predicted MoRFs in the 30–55; 95–115; and 135–155 regions. c-Myc also has three predicted MoRFs, residues 80–95; 200–220; and 330–350. n-Myc contains three predicted MoRFs at residues 30–50; 210–230; and 360–380. However, the shape of these dips corresponding to the predicted MoRFs and conserved regions is distorted in Myc proteins. In addition, conserved regions can also be found in the middle region at residues 235–240 and 280–285. Interestingly, these two conserved regions also correspond roughly to the locations of dips. They are located within long disordered regions. These long disordered regions are led or followed by several MoRFs/AiBSs.
![[K with combining low line]](https://www.rsc.org/images/entities/char_004b_0332.gif) ruppel-
ruppel-![[l with combining low line]](https://www.rsc.org/images/entities/char_006c_0332.gif) ike
ike ![[f with combining low line]](https://www.rsc.org/images/entities/char_0066_0332.gif) amily due to their homology to Drosophila melanogaster Kruppel protein. All Klf proteins are highly conserved at their C-terminal region by having three C2H2 zinc fingers and two H/C links.107 By taking Klf2 as an example, the three C2H2 zinc fingers are located at residues 273–295, 303–325, and 333–353. The two H/C links are TGEKPYH and TGHRPFQ, respectively. These regions are highly conserved among Klf2, Klf4, and Klf5. The highly conserved zinc fingers give Klf proteins very unique DNA binding properties, and all Klf proteins were shown to have very similar binding affinity on GC-rich regions and CACCC regions of DNA.108
amily due to their homology to Drosophila melanogaster Kruppel protein. All Klf proteins are highly conserved at their C-terminal region by having three C2H2 zinc fingers and two H/C links.107 By taking Klf2 as an example, the three C2H2 zinc fingers are located at residues 273–295, 303–325, and 333–353. The two H/C links are TGEKPYH and TGHRPFQ, respectively. These regions are highly conserved among Klf2, Klf4, and Klf5. The highly conserved zinc fingers give Klf proteins very unique DNA binding properties, and all Klf proteins were shown to have very similar binding affinity on GC-rich regions and CACCC regions of DNA.108
The disorder curves for Klf family proteins are shown in Fig. 5(c). All Klf proteins have three highly conserved C2H2 zinc finger domain at the very end of their C-termini starting from 410. Fragment 410–450 contains a MoRF in Klf4 corresponding to the first zinc finger and first H/C linker. Although there is no MoRF prediction in the same region of Klf2 and Klf5, the highly conserved sequence and similar disordered pattern may indicate the same function of this region among all Klf proteins. By taking the curve of Klf4 as the template, it is seen that many dips on Klf4 curve disappeared or became twisted in the curves of Klf2 and Klf5. Hence, identifying various interaction sites in this family could be very challenging. Actually, Klf4 has seven predicted MoRFs, while Klf5 has only three with two of them similar to those of Klf4. Klf2 has five predicted MoRFs. However, the locations and shapes of the disorder prediction curves associated with the Klf2 MoRFs are very different from the locations and shapes of the prediction curves associated with the Klf4 MoRFs.
Although Klf4 and Klf5 are generally more similar to each other, Klf4 has several regions highly similar to Klf2 instead of Klf5 (see also Sppl. Fig. 2). These regions in Klf4 are: 13–32; 95–112; and 384–400. By aligning only Klf4 and Klf2, it can be seen that the entire sequence of Klf 4 is highly similar to Klf2 with many insertions in Klf4. By comparing these insertions with alignment between Klf4 and Klf5, it was found that these insertions of Klf4 in the alignment of Klf4 and Klf2 are also insertions or less conserved segments in the alignment of Klf4 and Klf5. These alignments lead to very interesting conclusions on the functional evolution of Klf proteins. In addition to the highly conserved DNA-binding C-terminal regions, Klf4 has six predicted MoRFs. Four out of the six MoRFs are overlapped between Klf4 and Klf2 (ESI, Fig. S2‡). The fifth is in the insertion region of Klf4, while the last one is in the deletion region of Klf4. In the evolution of Klf2, the MoRF-indicated binding regions of Klf4 are maintained, while in the evolution of Klf5, the general sequence length and overall similarity are maintained.
evelopmental ![[p with combining low line]](https://www.rsc.org/images/entities/char_0070_0332.gif) luriotent-
luriotent-![[a with combining low line]](https://www.rsc.org/images/entities/char_0061_0332.gif) ssociated. This protein family has five members, of which four are associated with the cell pluripotency except Dppa1.8,90 Although this protein family is important in the development process, the structures and functions of its members are rarely studied especially on the protein level. Their sequence conservation is less strong (ESI, Fig. S1‡). Pair-wise alignments identified roughly two sub-groups of similar proteins: Dppa2 and Dppa4; Dppa3 and Dppa5 (ESI, Fig. S3‡). Proteins within each sub-group are similar to each other, although Dppa3 and Dppa5 do not have long conserved regions. The overall sequence identity and sequence similarity between Dppa3 and Dppa5 are 15% and 43%, respectively. Dppa2 and Dppa4 share three highly conserved regions: residues 85–120; 130–150; and 230–300 as shown in Fig. 5(d). The first two conserved regions contain a MoRF in both proteins. The third conserved region has much higher tendency to be ordered. However, both of the structure and function of this region are still unknown.
ssociated. This protein family has five members, of which four are associated with the cell pluripotency except Dppa1.8,90 Although this protein family is important in the development process, the structures and functions of its members are rarely studied especially on the protein level. Their sequence conservation is less strong (ESI, Fig. S1‡). Pair-wise alignments identified roughly two sub-groups of similar proteins: Dppa2 and Dppa4; Dppa3 and Dppa5 (ESI, Fig. S3‡). Proteins within each sub-group are similar to each other, although Dppa3 and Dppa5 do not have long conserved regions. The overall sequence identity and sequence similarity between Dppa3 and Dppa5 are 15% and 43%, respectively. Dppa2 and Dppa4 share three highly conserved regions: residues 85–120; 130–150; and 230–300 as shown in Fig. 5(d). The first two conserved regions contain a MoRF in both proteins. The third conserved region has much higher tendency to be ordered. However, both of the structure and function of this region are still unknown.
Dppa2 has a SAP domain at the position 85–119. SAP is a computationally identified conserved motif of about 35 residues involved in chromatin interaction. The conserved motif is named after AF-A/B, ![[A with combining low line]](https://www.rsc.org/images/entities/char_0041_0332.gif) cinus and ias.109 SAF-A/B is a nuclear protein called the caffold ttachment actors and
cinus and ias.109 SAF-A/B is a nuclear protein called the caffold ttachment actors and ![[B with combining low line]](https://www.rsc.org/images/entities/char_0042_0332.gif) . Acinus is a caspase-3-activated protein required for apoptotic chromatin condensation.110 Pias is a STAT inhibitor.111 As shown in Fig. 5(d), the SAP domain is predicted to have two MoRFs. Dppa4 also has a possible SAP domain due to similar sequences and highly conserved key residues of SAP domain shared by Dppa2 and Dppa4.112Dppa2 and Dppa4 further share a highly conserved segment of more than 70 residues at their C-termini. However, the function of this highly conserved region is still unknown.112 Another study on the Dppa2 mutation found that Dppa2-ΔN localized to the nucleus and induced two-cell-stage embryonic arrest.113 This result supports the transcriptional function of the SAP domain and the translocation function of its C-terminal region. Genomic analysis revealed that Dppa4 is only 16kb upstream of Dppa2.112 This observation could be the evidence that Dppa2 and Dppa4 are co-regulated. Actually, both of Dppa2 and Dppa4 are only expressed in pluripotent embryonic cells, but not in differentiated somatic cells. More interestingly, it was found that Oct4, Sox2, and Nanog can bind to Dppa4 promoter region in human ES cells.114
. Acinus is a caspase-3-activated protein required for apoptotic chromatin condensation.110 Pias is a STAT inhibitor.111 As shown in Fig. 5(d), the SAP domain is predicted to have two MoRFs. Dppa4 also has a possible SAP domain due to similar sequences and highly conserved key residues of SAP domain shared by Dppa2 and Dppa4.112Dppa2 and Dppa4 further share a highly conserved segment of more than 70 residues at their C-termini. However, the function of this highly conserved region is still unknown.112 Another study on the Dppa2 mutation found that Dppa2-ΔN localized to the nucleus and induced two-cell-stage embryonic arrest.113 This result supports the transcriptional function of the SAP domain and the translocation function of its C-terminal region. Genomic analysis revealed that Dppa4 is only 16kb upstream of Dppa2.112 This observation could be the evidence that Dppa2 and Dppa4 are co-regulated. Actually, both of Dppa2 and Dppa4 are only expressed in pluripotent embryonic cells, but not in differentiated somatic cells. More interestingly, it was found that Oct4, Sox2, and Nanog can bind to Dppa4 promoter region in human ES cells.114
Dppa3 is also called PGC7 (rimordial erm ell ![[7 with combining low line]](https://www.rsc.org/images/entities/char_0037_0332.gif) ). It is the first protein found in PGCs of both male and female. Dppa3 has a putative bipartite nuclear localization and export signal at around residues 30–60.115 This region is also suggested to be a putative SAP domain.116 In Fig. 5(d), this region of Dppa3 was predicted to be a MoRF. The region between residues 84 and 106 of Dppa3 is supposed to be a splicing factor domain.116 The location of the Dppa3 gene is in the conserved cluster containing Nanog94,117 and Gdf3.118 As shown in Table 1, both Nanog and Gdf3 are highly expressed in ES cells.
). It is the first protein found in PGCs of both male and female. Dppa3 has a putative bipartite nuclear localization and export signal at around residues 30–60.115 This region is also suggested to be a putative SAP domain.116 In Fig. 5(d), this region of Dppa3 was predicted to be a MoRF. The region between residues 84 and 106 of Dppa3 is supposed to be a splicing factor domain.116 The location of the Dppa3 gene is in the conserved cluster containing Nanog94,117 and Gdf3.118 As shown in Table 1, both Nanog and Gdf3 are highly expressed in ES cells.
Based on Pfam119 analysis, Dppa5 has a KH domain from residue 26 to residues 86. The KH domain is a K Homology domain, which was firstly identified in human heterogeneous nuclear ribonucleoprotein K. The primary function of the KH domain is RNA binding.120–122 There are two sub-types of KH domain: KH-1 and KH-2.123 The KH found in Dppa5 is a KH-1 domain, which has a βααββα structure.121 Prediction by Jpred124 on Dppa5 also shows this characteristic distribution on secondary structure elements (ESI, Fig. S4‡).
It is worth mentioning that Ecat1 also has a KH domain. The sequence alignment between Ecat1 and Dppa5 (ESI, Fig. S5‡) indicates their similarity within their KH domains. However, secondary structure prediction on Ecat1 by Jpred barely finds the first and the last helices of the βααββα fold (data not shown). Part of the reason for this result could be less conserved sequences of Ecat1 at the location of the first helix and two long insertions at the C-terminal region of KH domain of Ecat1.
Possible interaction sites within disordered regions identified by reverse alignment
Two sets of proteins, SOKM and SOLN, can induce the transformation of somatic sells into iPS cells. These two sets of proteins have two common reprogramming factors: Sox2 and Oct4. Further studies indicated that c-Myc and Lin28 are dispensable in each of the corresponding sets.10,13,81 Therefore, it can be assumed that Klf4 in SOKM and Nanog in SOLN may perform similar functions. However, the normal global sequence alignment between Klf4 and Nanog did not identify any significantly similar segments (ESI, Fig. S6‡). The local alignment identified two matched segments between these two proteins (ESI, Fig. S7‡). As shown in Fig. 6, these two matched segments are located in the regions with higher disorder scores, and their order is reversed within the corresponding sequences, suggesting that intrinsic disorder might play an important role in the spatial arrangement of protein interaction sites.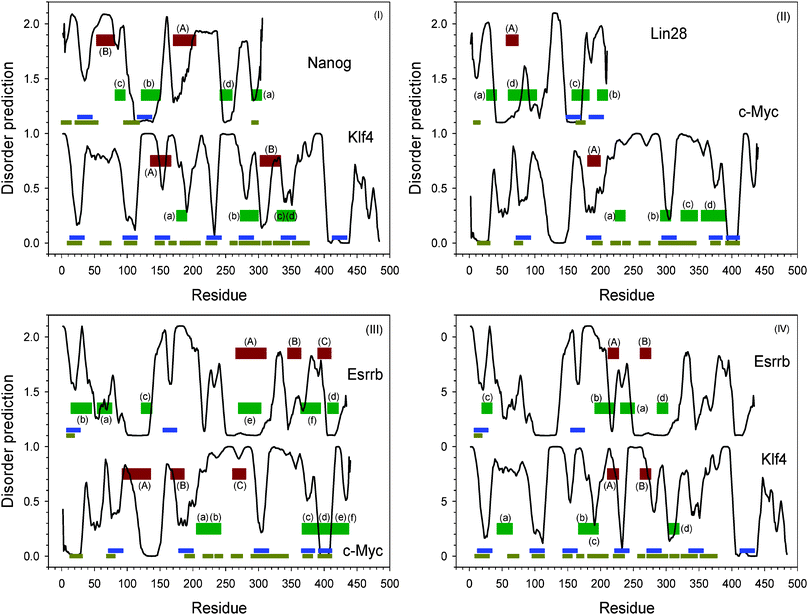 | ||
| Fig. 6 Similar segment identified by normal sequence alignment and reverse sequence alignment. (I) Klf4 and Nanog; (II) Lin28 and c-Myc; (III) Esrrb and Klf4; (IV) Esrrb and c-Myc. The black curves are intrinsic disorder prediction by PONDR®VL-XT. Blues bars are potential MoRF regions predicted by MoRF-II predictor.82 Horizontal dark yellow bars are binding motifs identified by ANCHOR.83,84 Dark red bars indicated by capital letters are matched segments by normal sequence alignment. Dark green shown together with lower-case letters bars are matched segments by reverse sequence alignment. Identical letters represent a pair of matched segment in sequence alignment. The details of the aligned sequences are shown in the ESI, Fig. S13.‡ | ||
In a few proven instances, protein binding domains are able to separately bind two different segments with the backbones oriented in opposite directions and with the two sequences containing similar amino acids in the reverse order. We conjecture that such oppositely oriented binding sites might be quite common, but are missed because no one looks for them. Based on this concept we constructed algorithms, called PONDR-RIBS, to identify reverse sequence alignments that could be indicators of oppositely directed binding sites to a common partner.125
By taking into consideration that both Klf4 and Nanog are highly disordered, a reverse sequence alignment was implemented by PONDR-RIBS.125 The results of this analysis are also presented in Fig. 6(I), which clearly shows that four other pairs of segments from the two proteins are identified to be reversely similar to each other. All the four pairs of segments correspond to dips in the disorder prediction plots of each protein. Segment (b), (c), and (d) of Klf4 are predicted to be MoRFs. The segment corresponding to (b) in Nanog is also predicted to be a MoRF.
It is likely that c-Myc and Lin28, being dispensable in SOKM and SOLN sets, also possess similar functions. As shown in Fig. 6(II), normal local sequence alignment identified one pair of similar segments in these two proteins. These segments were predicted to be a MoRF in c-Myc and a structure-prone motif before a flexible linker in Lin28. Reversed local sequence alignment with PONDR-RIBS125 discovered four additional pairs of reversely similar segments. In c-Myc, all these segments are located within a long disordered region. Segments (b) and (d) of c-Myc are also predicted to be MoRFs. In Lin28, segments (b) and (c) are predicted to be MoRFs; segment (a) is in a disordered region; and segment (d) expands a flexible linker in a structure-prone region.
Esrrb was reported be able to replace Klf4 and c-Myc.17 This is an indication that there could be some degree of sequence/function similarity among these three proteins. To show their similarities, the results from both normal local alignment and reversed local alignment are presented in Fig. 6(III). Esrrb and c-Myc have three similar segments. These three segments are in the same sequential order on both proteins. Segment (B) in c-Myc was identified as MoRF. Segments (A) are located in structure-prone regions of both proteins, whereas segments (C) appear as shallow dips. In the reversed alignment, six pairs of segments have similarity to each other. These six segments in c-Myc form two sequential clusters. Segments (a) and (b) are sequentially merged together, while the other four segments are also sequentially connected. The first cluster corresponds to small dips in disordered regions, while the second cluster contains several large dips in which (c) and (d) are predicted MoRFs. The corresponding reversed alignments of these six segments spread broadly in Esrrb. Segment (b) of Esrrb is a predicted MoRF. Segments (a), (d), and (f) are in various dips. Segments (c) and (e) are located within structure-prone regions.
Fig. 6(IV) shows that Esrrb and Klf4 possessed two pairs of normally aligned segments. The segments in Klf4 are overlapped with MoRF prediction. However, only segment (A) in Esrrb fits in a deep dip. Segment (B) of Esrrb is located within the structure-prone region. In the reversed alignment, four pairs of similar segments were identified. In Klf4, segment (a) is in disordered region. The other three are in dips. Especially, segment (b) and (c) are also mixed together. While in Esrrb, segment (a) and (d) are located inside the structure-prone regions; (b) and (c) stay in two dips. Segment (c) is also predicted as a MoRF.
Discussion
Many proteins are found closely related to the transformation of finally differentiated somatic cells into iPS cells. These proteins can be roughly classified into three categories: reprogramming factors that induce the iPS generation; proteins that influence the transformation efficiency; and proteins that are highly expressed during the transformation process. Here, we analyzed the sequence peculiarities and functional properties of various iPS-related proteins by using intrinsic disorder-based techniques such as intrinsic disorder prediction, MoRF prediction, and reverse sequence alignment. As indicated by our results, proteins functioning in iPS transformation are highly disordered. Intrinsic disorder-based bioinformatics analyses are powerful tools in revealing the functional roles of these proteins.The CH-plot used here was developed using known disordered proteins and known structured proteins, with the finding that the two groups are well separated by a straight line.30,74 There are complications to this simplified view suggesting structured proteins on one side of the boundary and disordered proteins on the other.
On the disordered protein side of the boundary, which contains very polar sequences, we envision at least 3 separate groups of sequences: positively charged, negatively charged, and neutral. The two differently charged sets are likely to be extended random coils, with the degree of extension being charge-dependent.126,127 The structural differences between the positively and negatively charged amino acids, however, means that the same distances from the CH boundary will likely lead to different characteristics for the positively and negatively charged disordered ensembles. Based on studies of simplified peptide polymers, neutral, but highly polar sequences are likely to be collapsed disordered structures.128,129
On the structured protein side of the boundary, two possibilities are evident. The original training set consisted of structured, globular, monomeric proteins, so proteins are likely to be of this type. A second possibility is that native molten globules would also be expected to be co-mingled with the structured proteins on the structured side of the boundary in the CH plot.
Further analysis and study are needed to sort out these various complications in the interpretation of the CH plot. Although difficult, such work is likely to bring additional insight into the structured and disordered characteristics of the protein factors that contribute to the development of iPS cells.
Many proteins are predicted to be structured by the CH plot and disordered by the CDF plot, thus falling into Q3. One possibility is that such proteins are molten globules. A second is that such proteins contain a mixture of structured and disordered regions. A combination of these two concepts, namely a protein having a secondary-structure-containing molten globular domain flanked by extended or collapsed disordered regions without secondary structure is also possible. Again, further analysis to sort out these alternatives would be useful to increase understanding of these transcription factors. These considerations point to further studies on these proteins.
The concept that transcription factors are mixtures of structure-forming modules, often undergoing disorder-to-order transitions, interspersed with regions of disorder has a long history. Brent and Ptashne130 demonstrated the feasibility of domain-swapping for these proteins; later, Ma and Ptashne131 found that for some transcription factors a high density of negative charges rather than a specific amino acid sequence was a major determinant of an activating region. From these and other reports, Sigler132 described transcription factors as “acid blobs” or “negative noodles.” By 1991, the disordered and modular status of many transcription factors was quite clear already.133
Once predictors of disorder were developed,30,72,73 an obvious application was to use such predictors to determine whether or not the disorder experimentally characterized for a few transcription factors132,133 is a general feature for these proteins. Such studies, carried out independently by two laboratories using completely different disordered predictors, yielded the similar results that almost every eukaryotic transcription factor is very rich in substantial-sized regions of disorder.26–28 A further interesting development has been to combine disorder prediction with information obtained from structural studies so that a fuller and richer understanding can be developed for each transcription factor.134 Here we are applying our own version135 of this last development to the various transcription factors that are important for iPS.
Disorder prediction-based analysis of the reprogramming factors is very helpful in identifying structural and even functional domains. As indicated by the results in Fig. 3, almost all the long order-prone regions identified by disorder prediction have PDB structures and are functional domains. This coincidence is actually a reflection of the relation between structure and function under the sequence-structure-function paradigm. However, beyond the order-prone regions, a lot of MoRFs were predicted in the intrinsically disordered regions of iPS-related proteins. The detailed binding mechanisms of these MoRFs interacting with their partners are still to be determined. A second predictor of binding sites, called ANCHOR, gave many predictions that overlapped with the MoRF predictions, but also a number of new potential sites of interaction.
While some of the predicted sites of interaction coincide with already determined binding sites, many of the predicted sites are not yet known to be associated with particular partners. Researchers have previously used such predictions of binding sites within disordered regions to help with the characterization of protein-protein interactions.136–138 Based on these previous successes, carrying out yeast-two hybrid or pull-down experiments with a focus on the predicted sites of interaction could provide new information regarding protein-protein interactions involving these factors. The results of such experiments could provide important information for understanding the mechanisms that underlie the conversion from somatic cells to iPS cells.
Within disordered regions, a number of so-called retro-MoRFs were identified by a combination of reversed sequence alignment and disorder prediction. In structured proteins, a segment has to adopt a specific conformation to properly interact with the other parts of the protein. If a segment is locally reversed, the structural details of the conformation formed by the reversed segment will be different from that of the original segment. Consequently, the reversed segment will be unlikely to fit into the structure formed by other parts of the structured protein. However, in disordered proteins or regions, the flexibility of disordered binding segments, or MoRFs, has been shown in some examples to enable reversed sequences to bind to the same partner but with the backbone pointing in the opposite direction. Therefore, when a segment within a disordered region is a MoRF, the reversed segment in another disordered region can potentially have the same binding function, but with a reversed backbone orientation. We called such segments retro-MoRFs.125 By applying the concepts of retro-MoRFs, new sites of potential functional similarity can be found. Since all the reprogramming factors are highly disordered and are enriched in the dips in their disorder predictions, and since many potential retro-MoRFs are identified in pairs of key proteins, it is conjectured that retro-MoRFs will be found to have important functional roles in the protein-protein and protein-DNA interactions that underlie the pathways leading to the development of iPS cells.
In addition to the MoRFs and retro-MoRFs, the reprogramming factors were shown to be enriched in binding regions identified by the ANCHOR program, regions that we have named AiBSs. Analysis revealed that MoRFs and AiBSs often overlapped. Furthermore, long AiBSs frequently contained several MoRFs and the overall patterns of the distribution of MoRFs and AiBSs within the sequences of members of a given family were rather conserved.
Although two groups of transcription factors have been proved to transform normal somatic cells into a status showing many properties of pluripotent cells, it is still unknown whether the iPS cells induced by these transcription factors have the complete set of normal biological functions as compared to ES cells. Our analysis indicates that these reprogramming transcription factors and many other transcription factors related to the iPS cells are highly disordered and have predictable functional properties. By taking into consideration that ES cell markers and reprogramming effectors are typically highly expressed at the reprogramming stage, it is likely that these additional transcription factors may also perform delicate functions in the reprogramming process. The functional involvement of these additional factors may be eventually responsible for the functional correctness of the generated iPS cells. The disordered analysis presented here, in our opinion, provides information that should be very useful in the further dissection of the structure/function relationships of these highly disordered proteins with their modular arrangements of functional domains.
Acknowledgements
This work was supported in part by the grant EF 0849803 from the National Science Foundation (to A.K.D and V.N.U.) and the Program of the Russian Academy of Sciences for the “Molecular and Cellular Biology” (to V.N.U.). We gratefully acknowledge the support of the IUPUI Signature Centers Initiative.References
- R. Jaenisch and R. Young, Cell, 2008, 132, 567–582 CrossRef CAS
.
- M. Kanatsu-Shinohara, K. Inoue, J. Lee, M. Yoshimoto, N. Ogonuki, H. Miki, S. Baba, T. Kato, Y. Kazuki, S. Toyokuni, M. Toyoshima, O. Niwa, M. Oshimura, T. Heike, T. Nakahata, F. Ishino, A. Ogura and T. Shinohara, Cell, 2004, 119, 1001–1012 CrossRef CAS
- M. Boiani and H. R. Scholer, Nat. Rev. Mol. Cell Biol., 2005, 6, 872–884 CrossRef CAS
- K. Okita, T. Ichisaka and S. Yamanaka, Nature, 2007, 448, 313–317 CrossRef CAS
- Y. F. Chou, H. H. Chen, M. Eijpe, A. Yabuuchi, J. G. Chenoweth, P. Tesar, J. Lu, R. D. McKay and N. Geijsen, Cell, 2008, 135, 449–461 CrossRef CAS
- J. T. Do and H. R. Scholer, Trends Pharmacol. Sci., 2009, 30, 296–302 CrossRef CAS
- I. H. Park, N. Arora, H. Huo, N. Maherali, T. Ahfeldt, A. Shimamura, M. W. Lensch, C. Cowan, K. Hochedlinger and G. Q. Daley, Cell, 2008, 134, 877–886 CrossRef CAS
- K. Takahashi and S. Yamanaka, Cell, 2006, 126, 663–676 CrossRef CAS
- K. Takahashi, K. Tanabe, M. Ohnuki, M. Narita, T. Ichisaka, K. Tomoda and S. Yamanaka, Cell, 2007, 131, 861–872 CrossRef CAS
- J. Yu, M. A. Vodyanik, K. Smuga-Otto, J. Antosiewicz-Bourget, J. L. Frane, S. Tian, J. Nie, G. A. Jonsdottir, V. Ruotti, R. Stewart, Slukvin II and J. A. Thomson, Science, 2007, 318, 1917–1920 CrossRef CAS
- V. Kashyap, N. C. Rezende, K. B. Scotland, S. M. Shaffer, J. L. Persson, L. J. Gudas and N. P. Mongan, Stem Cells Dev., 2009, 18, 1093–1108 CrossRef CAS
- J. L. Cox and A. Rizzino, Exp. Biol. Med., 2010, 235, 148–158 CrossRef CAS
- M. Nakagawa, M. Koyanagi, K. Tanabe, K. Takahashi, T. Ichisaka, T. Aoi, K. Okita, Y. Mochiduki, N. Takizawa and S. Yamanaka, Nat. Biotechnol., 2008, 26, 101–106 CrossRef CAS
- R. L. Judson, J. E. Babiarz, M. Venere and R. Blelloch, Nat. Biotechnol., 2009, 27, 459–461 CrossRef CAS
- D. Huangfu, K. Osafune, R. Maehr, W. Guo, A. Eijkelenboom, S. Chen, W. Muhlestein and D. A. Melton, Nat. Biotechnol., 2008, 26, 1269–1275 CrossRef CAS
- R. Blelloch, M. Venere, J. Yen and M. Ramalho-Santos, Cell Stem Cell, 2007, 1, 245–247 CrossRef CAS
- B. Feng, J. Jiang, P. Kraus, J. H. Ng, J. C. Heng, Y. S. Chan, L. P. Yaw, W. Zhang, Y. H. Loh, J. Han, V. B. Vega, V. Cacheux-Rataboul, B. Lim, T. Lufkin and H. H. Ng, Nat. Cell Biol., 2009, 11, 197–203 CrossRef CAS
- N. Tsubooka, T. Ichisaka, K. Okita, K. Takahashi, M. Nakagawa and S. Yamanaka, Genes Cells, 2009, 14, 683–694 CrossRef CAS
- A. Marson, R. Foreman, B. Chevalier, S. Bilodeau, M. Kahn, R. A. Young and R. Jaenisch, Cell Stem Cell, 2008, 3, 132–135 CrossRef CAS
- Y. Wang, K. Umeda and N. Nakayama, Stem Cell Res., 2010, 4, 223–231 CrossRef CAS
- R. M. Marion, K. Strati, H. Li, M. Murga, R. Blanco, S. Ortega, O. Fernandez-Capetillo, M. Serrano and M. A. Blasco, Nature, 2009, 460, 1149–1153 CrossRef CAS
- H. Hong, K. Takahashi, T. Ichisaka, T. Aoi, O. Kanagawa, M. Nakagawa, K. Okita and S. Yamanaka, Nature, 2009, 460, 1132–1135 CrossRef CAS
- T. Kawamura, J. Suzuki, Y. V. Wang, S. Menendez, L. B. Morera, A. Raya, G. M. Wahl and J. C. Belmonte, Nature, 2009, 460, 1140–1144 CrossRef CAS
- J. Utikal, J. M. Polo, M. Stadtfeld, N. Maherali, W. Kulalert, R. M. Walsh, A. Khalil, J. G. Rheinwald and K. Hochedlinger, Nature, 2009, 460, 1145–1148 CrossRef CAS
- J. Yu and J. A. Thomson, Genes Dev., 2008, 22, 1987–1997 CrossRef CAS
- J. Liu, N. B. Perumal, C. J. Oldfield, E. W. Su, V. N. Uversky and A. K. Dunker, Biochemistry, 2006, 45, 6873–6888 CrossRef CAS
- Y. Minezaki, K. Homma, A. R. Kinjo and K. Nishikawa, J. Mol. Biol., 2006, 359, 1137–1149 CrossRef CAS
- K. Nishikawa, Y. Minezaki and S. Fukuchi, Tanpakushitsu Kakusan Koso, 2006, 51, 1827–1835 CAS
- P. E. Wright and H. J. Dyson, J. Mol. Biol., 1999, 293, 321–331 CrossRef CAS
- V. N. Uversky, J. R. Gillespie and A. L. Fink, Proteins, 2000, 41, 415–427 CrossRef CAS
- A. K. Dunker, J. D. Lawson, C. J. Brown, R. M. Williams, P. Romero, J. S. Oh, C. J. Oldfield, A. M. Campen, C. M. Ratliff, K. W. Hipps, J. Ausio, M. S. Nissen, R. Reeves, C. Kang, C. R. Kissinger, R. W. Bailey, M. D. Griswold, W. Chiu, E. C. Garner and Z. Obradovic, J. Mol. Graphics Modell., 2001, 19, 26–59 CrossRef CAS
- P. Tompa, Trends Biochem. Sci., 2002, 27, 527–533 CrossRef CAS
- V. Receveur-Brechot, J. M. Bourhis, V. N. Uversky, B. Canard and S. Longhi, Proteins, 2006, 62, 24–45 CrossRef CAS
-
P. Tompa, Structure and function of intrinscially disordered proteins, Chapman & Hall/CRC Press, Boca Raton, 2009 Search PubMed
- P. E. Wright and H. J. Dyson, Curr. Opin. Struct. Biol., 2009, 19, 31–38 CrossRef CAS
- S. Longhi, Curr. Top. Microbiol. Immunol., 2009, 329, 103–128 CrossRef CAS
- A. K. Dunker, I. Silman, V. N. Uversky and J. L. Sussman, Curr. Opin. Struct. Biol., 2008, 18, 756–764 CrossRef CAS
- V. N. Uversky, C. J. Oldfield and A. K. Dunker, Annu. Rev. Biophys., 2008, 37, 215–246 CrossRef CAS
- V. N. Uversky, Protein J., 2009, 28, 305–325 CrossRef CAS
- V. N. Uversky and A. K. Dunker, Biochim. Biophys. Acta, Proteins Proteomics, 2010, 1804, 1231–1264 CrossRef CAS
- V. N. Uversky, Biochim. Biophys. Acta, Proteins Proteomics, 2011, 1814(5), 693–712 CrossRef CAS
- V. N. Uversky, Chem. Rev., 2011, 111, 1134–1166 CrossRef CAS
- B. He, K. Wang, Y. Liu, B. Xue, V. N. Uversky and A. K. Dunker, Cell Res., 2009, 19, 929–949 CrossRef CAS
- M. Fuxreiter, P. Tompa, I. Simon, V. N. Uversky, J. C. Hansen and F. J. Asturias, Nat. Chem. Biol., 2008, 4, 728–737 CrossRef CAS
- B. Ma and R. Nussinov, GenomeBiology, 2009, 10, 204 CrossRef
- M. R. Jensen, P. R. Markwick, S. Meier, C. Griesinger, M. Zweckstetter, S. Grzesiek, P. Bernado and M. Blackledge, Structure, 2009, 17, 1169–1185 CrossRef CAS
- A. Stein, R. A. Pache, P. Bernado, M. Pons and P. Aloy, FEBS J., 2009, 276, 5390–5405 CrossRef CAS
- R. Peters, Biochim. Biophys. Acta, Mol. Cell Res., 2009, 1793, 1533–1539 CrossRef CAS
- V. M. Navadgi-Patil and P. M. Burgers, DNA Repair, 2009, 8, 996–1003 CrossRef CAS
- J. A. Hebda and A. D. Miranker, Annu. Rev. Biophys., 2009, 38, 125–152 CrossRef CAS
- G. Harauz, V. Ladizhansky and J. M. Boggs, Biochemistry, 2009, 48, 8094–8104 CrossRef CAS
- J. Gsponer and M. M. Babu, Prog. Biophys. Mol. Biol., 2009, 99, 94–103 CrossRef CAS
- A. S. Garza, N. Ahmad and R. Kumar, Life Sci., 2009, 84, 189–193 CrossRef CAS
- C. A. Galea, Y. Wang, S. G. Sivakolundu and R. W. Kriwacki, Biochemistry, 2008, 47, 7598–7609 CrossRef CAS
- D. Eliezer, Curr. Opin. Struct. Biol., 2009, 19, 23–30 CrossRef CAS
- A. K. Dunker, C. J. Brown, J. D. Lawson, L. M. Iakoucheva and Z. Obradovic, Biochemistry, 2002, 41, 6573–6582 CrossRef CAS
- A. K. Dunker, C. J. Brown and Z. Obradovic, Adv. Protein Chem., 2002, 62, 25–49 CrossRef CAS
- J. J. Ward, J. S. Sodhi, L. J. McGuffin, B. F. Buxton and D. T. Jones, J. Mol. Biol., 2004, 337, 635–645 CrossRef CAS
- S. Vucetic, H. Xie, L. M. Iakoucheva, C. J. Oldfield, A. K. Dunker, Z. Obradovic and V. N. Uversky, J. Proteome Res., 2007, 6, 1899–1916 CrossRef CAS
- H. Xie, S. Vucetic, L. M. Iakoucheva, C. J. Oldfield, A. K. Dunker, Z. Obradovic and V. N. Uversky, J. Proteome Res., 2007, 6, 1917–1932 CrossRef CAS
- H. Xie, S. Vucetic, L. M. Iakoucheva, C. J. Oldfield, A. K. Dunker, V. N. Uversky and Z. Obradovic, J. Proteome Res., 2007, 6, 1882–1898 CrossRef CAS
- E. Hazy and P. Tompa, ChemPhysChem, 2009, 10, 1415–1419 CrossRef CAS
- R. B. Russell and T. J. Gibson, FEBS Lett., 2008, 582, 1271–1275 CrossRef CAS
- J. H. Han, S. Batey, A. A. Nickson, S. A. Teichmann and J. Clarke, Nat. Rev. Mol. Cell Biol., 2007, 8, 319–330 CrossRef CAS
- M. M. Pentony and D. T. Jones, Proteins: Struct., Funct., Bioinf., 2010, 78, 212–221 CrossRef CAS
- C. J. Brown, A. K. Johnson, A. K. Dunker and G. W. Daughdrill, Curr. Opin. Struct. Biol., 2011 Search PubMed
- C. J. Brown, S. Takayama, A. M. Campen, P. Vise, T. W. Marshall, C. J. Oldfield, C. J. Williams and A. K. Dunker, J. Mol. Evol., 2002, 55, 104–110 CrossRef CAS
- J. W. Chen, P. Romero, V. N. Uversky and A. K. Dunker, J. Proteome Res., 2006, 5, 888–898 CrossRef
- J. W. Chen, P. Romero, V. N. Uversky and A. K. Dunker, J. Proteome Res., 2006, 5, 879–887 CrossRef CAS
- P. Tompa, M. Fuxreiter, C. J. Oldfield, I. Simon, A. K. Dunker and V. N. Uversky, BioEssays, 2009, 31, 328–335 CrossRef CAS
- A. K. Dunker, Z. Obradovic, P. Romero, E. C. Garner and C. J. Brown, Genome Inform Ser Workshop Genome Inform, 2000, 11, 161–171 CAS
- P. Romero, Z. Obradovic, C. R. Kissinger, J. E. Villafranca and A. K. Dunker, IEEE Int. Conf. Neural Networks, 1997, 1, 90–95 CAS
- P. Romero, Z. Obradovic, X. Li, E. C. Garner, C. J. Brown and A. K. Dunker, Proteins: Struct., Funct., Genet., 2001, 42, 38–48 CrossRef CAS
- C. J. Oldfield, Y. Cheng, M. S. Cortese, C. J. Brown, V. N. Uversky and A. K. Dunker, Biochemistry, 2005, 44, 1989–2000 CrossRef CAS
- C. J. Oldfield, Y. Cheng, M. S. Cortese, P. Romero, V. N. Uversky and A. K. Dunker, Biochemistry, 2005, 44, 12454–12470 CrossRef CAS
- B. Xue, W. L. Hsu, J. H. Lee, H. Lu, A. K. Dunker and V. N. Uversky, Genes Cells, 2010, 15, 635–646 CrossRef CAS
- B. Xue, R. L. Dunbrack, R. W. Williams, A. K. Dunker and V. N. Uversky, Biochim. Biophys. Acta, Proteins Proteomics, 2010, 1804, 996–1010 CrossRef CAS
- P. V. Burra, L. Kalmar and P. Tompa, PLoS One, 2010, 5, e12069 Search PubMed
- Z. Obradovic, K. Peng, S. Vucetic, P. Radivojac, C. J. Brown and A. K. Dunker, Proteins: Struct., Funct., Genet., 2003, 53(Suppl 6), 566–572 CrossRef CAS
- T. Le Gall, P. R. Romero, M. S. Cortese, V. N. Uversky and A. K. Dunker, J. Biomol. Struct. Dyn., 2007, 24, 325–342 CAS
- M. Wernig, A. Meissner, J. P. Cassady and R. Jaenisch, Cell Stem Cell, 2008, 2, 10–12 CrossRef CAS
- Y. Cheng, C. J. Oldfield, J. Meng, P. Romero, V. N. Uversky and A. K. Dunker, Biochemistry, 2007, 46, 13468–13477 CrossRef CAS
- K. Peng, S. Vucetic, P. Radivojac, C. J. Brown, A. K. Dunker and Z. Obradovic, J. Bioinf. Comput. Biol., 2005, 3, 35–60 CrossRef CAS
- K. Peng, P. Radivojac, S. Vucetic, A. K. Dunker and Z. Obradovic, BMC Bioinformatics, 2006, 7, 208 CrossRef
- J. Prilusky, C. E. Felder, T. Zeev-Ben-Mordehai, E. H. Rydberg, O. Man, J. S. Beckmann, I. Silman and J. L. Sussman, Bioinformatics, 2005, 21, 3435–3438 CrossRef CAS
- Z. Dosztanyi, V. Csizmok, P. Tompa and I. Simon, Bioinformatics, 2005, 21, 3433–3434 CrossRef CAS
- A. Campen, R. M. Williams, C. J. Brown, J. Meng, V. N. Uversky and A. K. Dunker, Protein Pept. Lett., 2008, 15, 956–963 CrossRef CAS
- B. Meszaros, I. Simon and Z. Dosztanyi, PLoS Comput. Biol., 2009, 5, e1000376 Search PubMed
- Z. Dosztanyi, B. Meszaros and I. Simon, Bioinformatics, 2009, 25, 2745–2746 CrossRef CAS
- Z. Dosztanyi, V. Csizmok, P. Tompa and I. Simon, J. Mol. Biol., 2005, 347, 827–839 CrossRef CAS
- J. Kyte and R. F. Doolittle, J. Mol. Biol., 1982, 157, 105–132 CrossRef CAS
- B. Xue, C. J. Oldfield, A. K. Dunker and V. N. Uversky, FEBS Lett., 2009, 583, 1469–1474 CrossRef CAS
- A. Mohan, W. J. Sullivan Jr., P. Radivojac, A. K. Dunker and V. N. Uversky, Mol. BioSyst., 2008, 4, 328–340 RSC
- K. Mitsui, Y. Tokuzawa, H. Itoh, K. Segawa, M. Murakami, K. Takahashi, M. Maruyama, M. Maeda and S. Yamanaka, Cell, 2003, 113, 631–642 CrossRef CAS
- S. J. Elliman, I. Wu and D. M. Kemp, J. Biol. Chem., 2006, 281, 16–19 CrossRef CAS
- M. C. Wallis, P. D. Waters and J. A. Graves, Cell. Mol. Life Sci., 2008, 65, 3182–3195 CrossRef CAS
- J. O. Thomas, Biochem. Soc. Trans., 2001, 29, 395–401 CrossRef CAS
- M. Stros, D. Launholt and K. D. Grasser, Cell. Mol. Life Sci., 2007, 64, 2590–2606 CrossRef CAS
- V. Lefebvre, B. Dumitriu, A. Penzo-Mendez, Y. Han and B. Pallavi, Int. J. Biochem. Cell Biol., 2007, 39, 2195–2214 CrossRef CAS
- P. Tompa and M. Fuxreiter, Trends Biochem. Sci., 2008, 33, 2–8 CrossRef CAS
- S. I. Guth and M. Wegner, Cell. Mol. Life Sci., 2008, 65, 3000–3018 CrossRef CAS
- J. Gearhart, E. E. Pashos and M. K. Prasad, N. Engl. J. Med., 2007, 357, 1469–1472 CrossRef CAS
- R. Cotterman, V. X. Jin, S. R. Krig, J. M. Lemen, A. Wey, P. J. Farnham and P. S. Knoepfler, Cancer Res., 2008, 68, 9654–9662 CrossRef CAS
- J. Chaudhary and M. K. Skinner, Mol. Endocrinol., 1999, 13, 774–786 CrossRef CAS
- D. Dominguez-Sola, C. Y. Ying, C. Grandori, L. Ruggiero, B. Chen, M. Li, D. A. Galloway, W. Gu, J. Gautier and R. Dalla-Favera, Nature, 2007, 448, 445–451 CrossRef CAS
- W. H. Landschulz, P. F. Johnson and S. L. McKnight, Science, 1988, 240, 1759–1764 CAS
- S. V. Kozyrev, L. L. Hansen, A. B. Poltaraus, D. A. Domninsky and L. L. Kisselev, FEBS Lett., 1999, 448, 149–152 CrossRef CAS
- I. J. Miller and J. J. Bieker, Mol. Cell Biol., 1993, 13, 2776–2786 CAS
- L. Aravind and E. V. Koonin, Trends Biochem. Sci., 2000, 25, 112–114 CrossRef CAS
- S. Sahara, M. Aoto, Y. Eguchi, N. Imamoto, Y. Yoneda and Y. Tsujimoto, Nature, 1999, 401, 168–173 CrossRef CAS
- B. Liu, J. Liao, X. Rao, S. A. Kushner, C. D. Chung, D. D. Chang and K. Shuai, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 10626–10631 CrossRef CAS
- J. Maldonado-Saldivia, J. van den Bergen, M. Krouskos, M. Gilchrist, C. Lee, R. Li, A. H. Sinclair, M. A. Surani and P. S. Western, Stem Cells, 2007, 25, 19–28 CrossRef CAS
- J. Hu, F. Wang, X. Zhu, Y. Yuan, M. Ding and S. Gao, Dev. Dyn., 2010, 239, 407–424 CrossRef CAS
- L. A. Boyer, T. I. Lee, M. F. Cole, S. E. Johnstone, S. S. Levine, J. P. Zucker, M. G. Guenther, R. M. Kumar, H. L. Murray, R. G. Jenner, D. K. Gifford, D. A. Melton, R. Jaenisch and R. A. Young, Cell, 2005, 122, 947–956 CrossRef CAS
- M. Sato, T. Kimura, K. Kurokawa, Y. Fujita, K. Abe, M. Masuhara, T. Yasunaga, A. Ryo, M. Yamamoto and T. Nakano, Mech. Dev., 2002, 113, 91–94 CrossRef CAS
- B. Payer, M. Saitou, S. C. Barton, R. Thresher, J. P. Dixon, D. Zahn, W. H. Colledge, M. B. Carlton, T. Nakano and M. A. Surani, Curr. Biol., 2003, 13, 2110–2117 CrossRef CAS
- I. Chambers, D. Colby, M. Robertson, J. Nichols, S. Lee, S. Tweedie and A. Smith, Cell, 2003, 113, 643–655 CrossRef CAS
- A. A. Caricasole, R. H. van Schaik, L. M. Zeinstra, C. D. Wierikx, R. J. van Gurp, M. van den Pol, L. H. Looijenga, J. W. Oosterhuis, M. F. Pera, A. Ward, D. de Bruijn, P. Kramer, F. H. de Jong and A. J. van den Eijnden-van Raaij, Oncogene, 1998, 16, 95–103 CAS
- R. D. Finn, J. Mistry, J. Tate, P. Coggill, A. Heger, J. E. Pollington, O. L. Gavin, P. Gunasekaran, G. Ceric, K. Forslund, L. Holm, E. L. Sonnhammer, S. R. Eddy and A. Bateman, Nucleic Acids
Res., 2009, 38, D211–222 CrossRef
- C. G. Burd and G. Dreyfuss, Science, 1994, 265, 615–621 CAS
- G. Musco, A. Kharrat, G. Stier, F. Fraternali, T. J. Gibson, M. Nilges and A. Pastore, Nat. Struct. Biol., 1997, 4, 712–716 CrossRef CAS
- J. L. Baber, D. Libutti, D. Levens and N. Tjandra, J. Mol. Biol., 1999, 289, 949–962 CrossRef CAS
- N. V. Grishin, Nucleic Acids Res., 2001, 29, 638–643 CrossRef CAS
- C. Cole, J. D. Barber and G. J. Barton, Nucleic Acids Res., 2008, 36, W197–201 CrossRef CAS
- B. Xue, A. K. Dunker and V. N. Uversky, Int. J. Mol. Sci., 2010, 11, 3725–3747 CrossRef CAS
- J. Yamada, J. L. Phillips, S. Patel, G. Goldfien, A. Calestagne-Morelli, H. Huang, R. Reza, J. Acheson, V. V. Krishnan, S. Newsam, A. Gopinathan, E. Y. Lau, M. E. Colvin, V. N. Uversky and M. F. Rexach, Mol. Cell. Proteomics, 2010, 9, 2205–2224 CAS
- A. H. Mao, S. L. Crick, A. Vitalis, C. L. Chicoine and R. V. Pappu, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 8183–8188 CrossRef CAS
- S. L. Crick, M. Jayaraman, C. Frieden, R. Wetzel and R. V. Pappu, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 16764–16769 CrossRef CAS
- H. T. Tran, A. Mao and R. V. Pappu, J. Am. Chem. Soc., 2008, 130, 7380–7392 CrossRef CAS
- R. Brent and M. Ptashne, Cell, 1985, 43, 729–736 CrossRef CAS
- J. Ma and M. Ptashne, Cell, 1987, 51, 113–119 CrossRef CAS
- P. B. Sigler, Nature, 1988, 333, 210–212 CrossRef CAS
- A. D. Frankel and P. S. Kim, Cell, 1991, 65, 717–719 CrossRef CAS
- S. Fukuchi, K. Homma, Y. Minezaki, T. Gojobori and K. Nishikawa, BMC Struct. Biol., 2009, 9, 26 CrossRef
- L. M. Iakoucheva, C. J. Brown, J. D. Lawson, Z. Obradovic and A. K. Dunker, J. Mol. Biol., 2002, 323, 573–584 CrossRef CAS
- A. J. Callaghan, J. P. Aurikko, L. L. Ilag, J. Gunter Grossmann, V. Chandran, K. Kuhnel, L. Poljak, A. J. Carpousis, C. V. Robinson, M. F. Symmons and B. F. Luisi, J. Mol. Biol., 2004, 340, 965–979 CrossRef CAS
- S. Longhi, V. Receveur-Brechot, D. Karlin, K. Johansson, H. Darbon, D. Bhella, R. Yeo, S. Finet and B. Canard, J. Biol. Chem., 2003, 278, 18638–18648 CrossRef CAS
- J. C. Rosenbaum, E. K. Fredrickson, M. L. Oeser, C. M. Garrett-Engele, M. N. Locke, L. A. Richardson, Z. W. Nelson, E. D. Hetrick, T. I. Milac, D. E. Gottschling and R. G. Gardner, Mol. Cell, 2011, 41, 93–106 CrossRef CAS
Footnotes |
| † Published as part of a Molecular BioSystems themed issue on Intrinsically Disordered Proteins: Guest Editor M Madan Babu. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c1mb05163f |
| This journal is © The Royal Society of Chemistry 2012 |