An electrochemical gas sensor based on paper supported room temperature ionic liquids
Nicolò
Dossi
a,
Rosanna
Toniolo
*a,
Andrea
Pizzariello
a,
Emanuel
Carrilho
bc,
Evandro
Piccin
d,
Simone
Battiston
e and
Gino
Bontempelli
a
aDepartment of Food Science, University of Udine, via Cotonificio 108, I-33100, Udine, Italy. E-mail: rosanna.toniolo@uniud.it; Fax: +39 0432-558803; Tel: +39 0432-558885
bInstitute of Chemistry at São Carlos, University of São Paulo, 13566-590, São Carlos, SP, Brazil
cNational Institute of Bioanalytical Science and Technology, 13083-901, Campinas, SP, Brazil
dDepartment of Chemistry, Federal University of Minas Gerais, 31270-901, Belo Horizonte, MG, Brazil
eCNR Institute for Energetics and Interphases, Corso Stati Uniti 4, I-35127, Padova, Italy
First published on 10th November 2011
Abstract
A sensitive and fast-responding membrane-free amperometric gas sensor is described, consisting of a small filter paper foil soaked with a room temperature ionic liquid (RTIL), upon which three electrodes are screen printed with carbon ink, using a suitable mask. It takes advantage of the high electrical conductivity and negligible vapour pressure of RTILs as well as their easy immobilization into a porous and inexpensive supporting material such as paper. Moreover, thanks to a careful control of the preparation procedure, a very close contact between the RTIL and electrode material can be achieved so as to allow gaseous analytes to undergo charge transfer just as soon as they reach the three-phase sites where the electrode material, paper supported RTIL and gas phase meet. Thus, the adverse effect on recorded currents of slow steps such as analyte diffusion and dissolution in a solvent is avoided. To evaluate the performance of this device, it was used as a wall-jet amperometric detector for flow injection analysis of 1-butanethiol vapours, adopted as the model gaseous analyte, present in headspace samples in equilibrium with aqueous solutions at controlled concentrations. With this purpose, the RTIL soaked paper electrochemical detector (RTIL-PED) was assembled by using 1-butyl-3-methylimidazolium bis(trifluoromethanesulfonyl)imide as the wicking RTIL and printing the working electrode with carbon ink doped with cobalt(II) phthalocyanine, to profit from its ability to electrocatalyze thiol oxidation. The results obtained were quite satisfactory (detection limit: 0.5 μM; dynamic range: 2–200 μM, both referring to solution concentrations; correlation coefficient: 0.998; repeatability: ±7% RSD; long-term stability: 9%), thus suggesting the possible use of this device for manifold applications.
1. Introduction
Nowadays, there is a strong demand for highly selective, sensitive, stable and cost-effective gas sensors suitable for environmental monitoring,1 clinical diagnosis2 and food quality control.3Electroanalytical sensors offer quite attractive online capabilities and in situ multicomponent measurements, almost always directly in samples in which the analytes of interest are present, with low-cost instrumentation. Unfortunately, usual amperometric devices cannot be applied directly to gaseous samples in which no supporting electrolyte is present and neither can be added. To overcome this inconvenience, gas-permeable membrane electrodes were developed, based on permeation of gaseous analytes through a membrane which dissolve in an internal electrolyte solution and diffuse to the working electrode. Their performance is conditioned by these slow steps because they cause lowering of sensitivity and lengthening of response time.4,5 Moreover, the electrolyte solvent can evaporate making these devices fail. To avoid these drawbacks, amperometric gas sensors based on the use of moist ion-exchange membranes as solid polymer electrolytes (SPEs) were developed in the last two decades.4–13 In these devices the membrane separating the gaseous samples from the internal electrolyte is not permeated by analytes but serves to provide the transfer of charged species from the working to the counter electrode, thus playing the role assumed by usual supporting electrolytes. Remarkable benefits are gained from the use of this assembly thanks to the elimination of a permeation step.14 But these electrodes are also prone to drying out, even though to a lesser extent with respect to permeation sensors.
These problems could be conceivably avoided by replacing conventional electrochemical solvents with room temperature ionic liquids (RTILs). These low melting salts display large electrochemical windows, negligible vapour pressure, good thermal stability and electrical conductivity which make them quite attractive. In fact, some amperometric gas sensors were recently proposed which were assembled by casting a thin layer of RTIL on the surface of either a three electrode cell or a microelectrode array.15–17 Even though these sensors are able to operate as membrane-free amperometric devices, their responses remain conditioned by the rate of both gas dissolution into the RTIL and its diffusion through the medium towards the working electrode. Thus, their sensitivity and response time continue to be affected by fairly slow steps, even though they are faster compared to permeation through membranes. Moreover, variations of gas solubility and diffusion rate with the temperature can affect their responses significantly. Consequently, the design and development of a new class of RTIL based sensors is highly desirable. In particular, it is advisable to achieve a very close contact between the electrode material and RTIL, thus allowing analytes from gaseous samples to undergo electron transfer just as they reach the working-electrode-material/RTIL interphase, without involving any analyte diffusion and/or dissolution step. Concomitantly, the RTIL medium available in close contact with the electrode material can ensure the transfer of charged species from the working electrode to the counter electrode. Thus, the aim of this investigation was the development of RTIL based membrane-free and fast-responding amperometric gas sensors able to provide highly sensitive responses thanks to this strategy.
To achieve this goal, conventional filter paper was used to support both the RTIL and electrodes which were suitably screen printed with carbon ink. This type of support was chosen in view of its large availability at low cost, high porosity and hydrophilic properties, as well as its suitability to undergo screen printing processes. On the other hand, this material was extensively used in the past as a platform for analytical and clinical chemistry applications.18,19 Moreover, paper was also adopted for assembling cheap microfluidic devices consisting of a set of hydrophilic capillary channels whose boundaries were defined by hydrophobic barriers prepared by either photolithography or inkjet and wax printing.20–23 In particular, some microfluidic paper based devices were proposed recently which were directly integrated with colorimetric,24 chemiluminescence25,26 or electrochemical27–29detectors.
The paper-RTIL based electrochemical gas sensor here proposed consists of a carbon ink screen printed miniaturized three electrode cell defined by a circle of hydrophobic wax barrier. Its performance was evaluated by monitoring oxidation currents for 1-butanethiol, as the model electroactive gaseous analyte, recorded either directly or mediated by the cobalt(II) phthalocyanine complex, acting as an electrocatalyst30 immobilized onto the electrode surface.
2. Experimental section
2.1 Chemicals and instrumentation
All the chemicals used were of analytical reagent grade quality and were employed as received, without further purification. 1-Butyl-3-methylimidazolium bis(trifluoromethanesulfonyl)imide [BMIM][NTF2], 1-butyl-3-methylimidazolium hexafluorophosphate [BMIM][PF6], 1-butyl-3-methylimidazolium tetrafluoroborate [BMIM][BF4], Co(II) phthalocyanine, ferrocene, phenol, 1-butanethiol, acetone and cyclohexanone were purchased from Sigma-Aldrich (St Louis, MO, USA). Filter paper type 1 and conductive carbon powder (325 mesh) were obtained from Whatman (Maidstone, UK) and Alfa Aesar (Ward Hill, MA, USA), respectively.The carbon ink was prepared in-house by suspending 0.5 g of graphite powder in 1 mL of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 acetone–cyclohexanone mixture in which 0.08 g of PVC were dissolved.31Carbon ink chemically modified with Co(II) phthalocyanine was prepared by dissolving 25 mg of the Co complex in this suspension.32 Microcircuits were printed on filter paper with a Xerox Phaser 8560 N wax ink printer.
:1 acetone–cyclohexanone mixture in which 0.08 g of PVC were dissolved.31Carbon ink chemically modified with Co(II) phthalocyanine was prepared by dissolving 25 mg of the Co complex in this suspension.32 Microcircuits were printed on filter paper with a Xerox Phaser 8560 N wax ink printer.
All voltammetric and amperometric measurements were performed using a PalmSens electrochemical analyser (Palm Instruments, Houten, The Netherlands) driven by a software installed on a Pentium IV computer. Flow injection analyses were carried out by using ultrapure nitrogen as carrier gas purchased from SIAD (Trieste, Italy), whose flow rate was controlled in the range of 20–100 mL min−1 by a micrometric valve (Viton SS-22RS2).
2.2 Preparation of RTIL-paper based sensors
Wax printing was used to pattern filter paper according to a previously reported method.22 Briefly, a series of rings (6 mm i.d., with a line thickness of 2 mm) were printed with black wax-based ink onto a filter paper foil to define the area of a set of sensors. Heating at 120 °C for 10 min was adopted to melt the wax into the printed paper which was then cut into pieces, each containing a single circular pad displaying a hydrophilic paper area of 28.14 mm2, defined by a hydrophobic barrier. The back face of this patterned paper was insulated by thermally laminating a polyethylene (PET) layer (0.1 mm) to prevent any electrolyte leakage and gas permeation during analysis. Subsequently, reference, counter and working (1.3 × 4.0 mm) electrodes were screen printed on the top face of the device with carbon ink, exploiting a suitable mask drawn by the Freehand 7.0 software (Macromedia, San Francisco, CA, USA). The same procedure was adopted to print working electrodes chemically modified with Co(II) phthalocyanine. Electrical connections to all electrodes were located outside the circular hydrophobic wax barriers surrounding hydrophilic cells. Finally, a controlled volume of RTIL (1.7 μL) was gently laid on a corner of the paper device in order to soak in paper channels, without covering the upper surface of electrodes.The layout of this RTIL soaked paper electrochemical detector (RTIL-PED) is shown in Fig. 1.
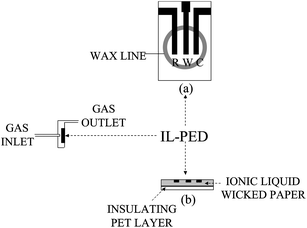 | ||
| Fig. 1 Layout (a) and cross-section (b) of the RTIL-PED sensor adopted in flow injection analyses. R, pseudo-reference electrode; W, working electrode; C, counter electrode. | ||
2.3 Electrochemical and morphological measurements
Voltammetric experiments were performed in both RTIL solution and gas phase. A conventional three-electrode cell (2 mL) was adopted for conducting voltammetric tests in solution. Its Teflon cover was provided with three holes for inserting a platinum wire counter electrode, an Ag/AgCl reference electrode and a glassy carbon disk working electrode, whose surface was mirror-polished with fine alumina powder prior to each set of experiments.Voltammetric measurements in gas phase were instead performed by tightly piercing a RTIL-PED in the stopper of a 10 mL plastic vial (2 cm external diameter; 3 cm in height), keeping outside electrical connections. The RTIL-PED was exposed to the headspace equilibrated with analyte vapours (1-butanethiol or phenol) released from solutions (1 mL) at controlled concentrations in either RTIL or water, which were kept at room temperature and stirred at a constant rate (100 rev min−1) with an Amel 291 Mast magnetic stirrer (Milan, Italy). Voltammograms were typically recorded at a scan rate of 50 mV s−1.
The potential of the pseudo-reference electrode was periodically monitored by dissolving in the RTIL medium small amounts of ferrocene (10 mM) whose potential was previously evaluated vs. an Ag/AgCl, Cl−satelectrode. In this way, a potential of ca. 120 mV vs.Ag/AgCl, Clsat could be estimated for the pseudo-reference carbon ink electrode.
Flow injection amperometric measurements were carried out with a home-made flow apparatus consisting of a thin stainless steel tubing (60 cm long; 0.5 mm i.d.) half of which was rolled up to act as a pulse damper. It was fed with nitrogen at a controlled flow rate (10–100 mL min−1), monitored by a flowmeter inserted in the stream. This apparatus was equipped with an air-thermostatted injection device made with a rubber cup through which controlled headspace volumes (0.2–2.0 mL) were injected with gas-tight syringes. The outlet of the stainless steel tubing was positioned in front of the RTIL-PED sensor, in a wall-jet configuration, at a distance, controlled by a plastic spacer, of 1 mm from its surface. This sensor was housed in a small PET container, provided with a gas outlet and an inlet port, the latter connected to the outlet of the stainless steel tubing. It was fixed on an inner wall by using simple double side adhesive tape. A detection potential of 2.2 or 1.6 V was applied to the working electrode, depending upon whether it consisted of unmodified or chemically modified carbon ink, respectively. Sample injections were performed after rapid baseline stabilization and current signals were sampled with a time resolution of 0.05 s.
The main operative parameters for this type of measurement were preliminarily optimised by carrying out some tests on purpose. They led to infer that the best results were achieved by injecting 1.0 mL of gaseous sample, using a carrier flow rate of 50 mL min−1 and adopting a distance of 1 mm between the sensing surface and the sample outlet.
Morphological characterization of the surface of RTIL-PED sensors was performed by scanning electron microscope-energy dispersion spectroscopic (SEM-EDS) analyses. Energy Dispersive Spectroscopy (EDS, Oxford X-Max) compositional maps of the RTIL-PED sensor surfaces were collected by a Field Emission Scanning Electron Microscope (Sigma FESEM, Carl Zeiss—Oberkochen, Germany) at an acceleration voltage of 15 kV. The observed samples were coated with platinum by an Emitech sputter coater K575X (Emitech—Ashford, UK).
3. Results and discussion
3.1 Morphological and voltammetric characterization of RTIL-paper based sensors
Some SEM-EDS and voltammetric tests were preliminarily performed to identify the best RTIL for assembling RTIL-PEDs. With this purpose, sensors were prepared by using alternatively [BMIM][NTF2], [BMIM][BF4] or [BMIM][PF6] as the wicking RTIL, they being chosen on the basis of their wide electrostability range.16 Thus, about 1.7 μL of each RTIL (equivalent to ca. 5.7 μmol of [BMIM][NTF2], 9.1 μmol of [BMIM][BF4] and 8.1 μmol of [BMIM][PF6], on the basis of their molecular weight and density, the density being 1.44, 1.21 and 1.37 g cm−3, respectively)33 were applied to soak the hydrophilic paper area in different pads where electrodes were preliminarily screen printed. Subsequently, these PEDs were periodically subjected to both SEM-EDS and voltammetric monitoring.SEM analyses of PED surfaces showed that a homogeneous distribution of [BMIM][NTF2] was quite rapidly achieved (in about 5 min), while a not completely homogeneous distribution of [BMIM][BF4] was attained even after rather longer times. [BMIM][PF6] turned out to be quite reluctant to fill paper pores. In other words, the ability of the tested RTILs to soak paper turned out to depend markedly on their viscosity, whose values are 52, 112 and 371 cP, respectively.33 These findings agree well with the results reported in a previous investigation concerning hydrophilic nylon membranes impregnated with the same RTILs.34SEM micrographs also showed that the amount of RTIL used to wet paper was just suitable for avoiding its overflow over the carbon-ink surface, thus preventing the undesired formation of a thin RTIL film covering the working electrode surface.
To achieve a deeper insight into the RTIL–carbon ink interphase, SEM-EDS micrographs were recorded on the three types of RTIL-PEDs at the energy value (0.677 keV) proper for the fluorine peak, present in all the RTILs assayed. The results obtained are summarized in Fig. 2A which shows that [BMIM][NTF2] made it possible to attain the best composites. In fact, high amounts of this RTIL, homogeneously distributed and in close contact with carbon particles turned out to enter paper pores in the portion coated by printed electrodes (maps on the left hand side of Fig. 2). Instead, this carbon coated paper portion appeared to be wicked by quite lower amounts of other RTILs.
![(A) Fluorine Kα1,2 EDS maps (0.677 KeV) of PED surfaces prepared by using paper soaked with: (a) [BMIM][NTF2]; (b) [BMIM][BF4]; (c) [BMIM][PF6] and (B) the corresponding cyclic voltammograms recorded at 50 mV s−1 when they were exposed to headspaces equilibrated with: pure water (dotted lines) and a 10 mM aqueous solution of phenol (full lines).](/image/article/2012/LC/c1lc20663j/c1lc20663j-f2.gif) | ||
| Fig. 2 (A) Fluorine Kα1,2 EDS maps (0.677 KeV) of PED surfaces prepared by using paper soaked with: (a) [BMIM][NTF2]; (b) [BMIM][BF4]; (c) [BMIM][PF6] and (B) the corresponding cyclic voltammograms recorded at 50 mV s−1 when they were exposed to headspaces equilibrated with: pure water (dotted lines) and a 10 mM aqueous solution of phenol (full lines). | ||
A confirmation for this evidence was gained by recording cyclic voltammograms at these RTIL-PEDs exposed to the headspace equilibrated with either pure water or 10 mM aqueous solutions of phenol. As shown in Fig. 2B, both background currents and peak currents for phenol decreased markedly on passing from [BMIM][NTF2] to [BMIM][BF4] and, further, to [BMIM][PF6], in agreement with a decrease of the effective area of carbon particles contacted by the wicking RTIL. The use of [BMIM][NTF2] turned out to be preferable also because exposure of these RTIL-PEDs to water vapors for fairly long times (some tens of min) caused a progressive, even though poorly marked, narrowing of the available potential windows which was more marked for RTIL-PEDs prepared by using [BMIM][BF4] and [BMIM][PF6], in agreement with their stronger affinity with water.33
On the basis of these findings, all the subsequent investigations were conducted with [BMIM][NTF2]-soaked RTIL-PEDs.
3.2 Voltammetric behaviour of 1-butanethiol vapours at RTIL-PEDs
In order to define the best conditions for the detection of 1-butanethiol, adopted by us as the model gaseous analyte, linear sweep voltammograms for this species were preliminarily recorded at both glassy carbon electrodes in RTIL solutions and RTIL-PEDs exposed to headspaces equilibrated with analyte vapours released from aqueous solutions at controlled concentrations.1-Butanethiol dissolved in [BMIM][NTF2] displayed at glassy carbon electrodes a well defined sharp anodic peak at about 1.5 V, clearly separated from the solvent discharge (about 2.1 V). When linear sweep voltammograms were instead recorded at the RTIL-SPE sensor, the anodic process for 1-butanethiol vapours occurred at more anodic potentials, close to the solvent discharge, so that these oxidations turned out to be almost indistinguishable from each other, as shown in Fig. 3A. This was not surprising since a marked overpotential increase is frequently encountered for several electrochemical processes when glassy carbon surfaces are replaced by carbon paste materials. This is because even weaker adsorption effects involving electroactive species, intermediates or products are greatly enhanced at these latter large-surface electrodes, thus affecting significantly electron transfer rates and/or modifying to some extent the reaction pathway involved in the electrochemical process.
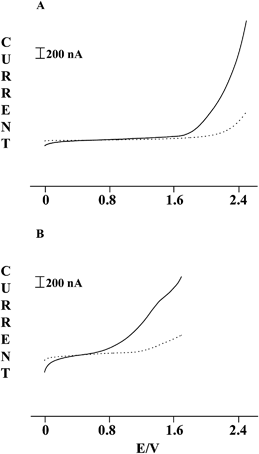 | ||
| Fig. 3 Linear sweep voltammograms recorded with a scan rate of 50 mV s−1 at RTIL-PEDs prepared by screen printing the working electrode with: (A) simple carbon ink; (B) carbon ink containing 5% w/w Co(II) phthalocyanine. Dotted lines refer to headspaces equilibrated with pure water, while full lines report the profiles found for a 3 mM aqueous solution of 1-butanethiol. | ||
The occurrence of 1-butanethiol oxidation at potentials close to the solvent discharge prevented its reliable monitoring by RTIL-PEDs prepared with simple carbon ink. To overcome this drawback, suitable amounts of Co(II) phthalocyanine were added to the carbon ink used for screen printing the working electrode, to profit from the well known electrocatalytic process taking place when thiol oxidation is conducted in the presence of this complex.30 In fact, this process, occurring through the following reaction pathway, allowed the potential required for thiol oxidation to be markedly lowered:
| [Co(II)-phthal] → [Co(III)-phthal]+ + e− | (1) |
| [Co(III)-phthal]+ + RSH → RS˙ + H+ + [Co(II)-phthal] | (2) |
| 2 RS˙ → RSSR | (3) |
Unfortunately, the scarce solubility of Co(II) phthalocyanine in RTILs prevented us from checking that its electrocatalytic effect on thiol oxidation is also operative in [BMIM][NTF2] by carrying out simple voltammetric tests in RTIL solutions containing 1-butanethiol together with the mentioned Co complex. Consequently, this check was carried out by recording voltammetric tests in gas phase at chemically modified (CM) RTIL-PEDs. When these CM-RTIL-PEDs were exposed to headspaces equilibrated with pure water, an anodic wave very poorly defined and scarcely distinguishable from the background current was observed at about 1.4 V for the oxidation of Co(II) to Co(III). Nevertheless, a remarkable increase of this wave was observed when the sensor was exposed to headspaces equilibrated with 1-butanethiol solutions, as shown in Fig. 3B. In particular, the relevant anodic current turned out to increase linearly in a rather wide range (0.01–100 mM) with the 1-butanethiol concentration in aqueous solutions.
3.3 Flow injection analysis at CM-RTIL-PEDs
These analyses were conducted using RTIL-PEDs chemically modified by Co(II)-phthalocyanine as wall-jet amperometric detectors in the flow apparatus described in Section 2.3, under the optimised conditions reported in Section 2.3. Aqueous solutions containing 1-butanethiol at different concentrations (2–200 μM) were prepared and thermostatted at room temperature. Controlled headspace volumes (1.0 mL) were then injected with gas-tight syringes and conveyed by the nitrogen carrier gas (flow rate 50 mL min−1) to the CM-RTIL-PED to which a potential of 1.6 V was applied.Detection of 1-butanethiol by flow injection resulted in sharp peak readouts with rapid increases and decreases of the current that reflected the passage of the sample zone over the working electrode and which were superimposed on a rather flat baseline. Fig. 4 reports some typical responses obtained. The height of recorded peaks depended linearly on 1-butanethiol solution concentration over a wide range (2–200 μM) with the following regression equation: i (nA) = 54.52C (μM) − 3.35 and a good correlation coefficient (r = 0.998). It was also characterized by a satisfactory repeatability (ca. 7% RSD), which was estimated, on average, for peaks recorded for seven replicate measurements on the same samples, independently of their analyte content. From the slope of the mentioned calibration plot, a detection limit of 0.5 μM could be inferred for a signal-to-noise ratio of 3. Moreover, in order to roughly estimate the long-term stability of the amperometric sensor, current signals were recorded in replicate injections (n = 7) of headspace samples equilibrated with different thiol concentrations which were repeated every day for three weeks. On average, the signal decreased by about 9% on passing from the first to the last day. Finally, inter-electrode reproducibility was estimated by testing with different RTIL-PEDs, all prepared by the method in Section 2.2, the same 1-butanethiol samples. They led to quite similar responses (±9%), thus indicating that active electrode surfaces reproducible enough can be achieved by the procedure suggested here.
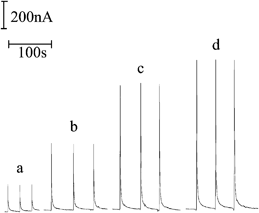 | ||
| Fig. 4 Flow injection peaks recorded at a RTIL-PED chemically modified with Co(II) phthalocyanine for headspaces in equilibrium with aqueous solutions containing 1-butanethiol at the following concentrations: (a) 3.8 μM; (b) 8.5 μM; (c)16.0 μM; (d) 20.0 μM. Electrode potential: 1.6 V; carrier gas flow rate: 50 mL min−1; sample volume: 1 mL; distance of sample outlet from sensing surface: 1 mm. | ||
The fast response achieved in these flow injection analyses (base peak width < 6 s) shows evidence for the fact that this approach allows an injection frequency even higher than 200 per hour. This profitable throughput comes from the spread and close contact between electrode material and RTIL electrolyte, enabling gaseous analytes to undergo charge transfer just as they reach the three-phase sites where electrode particles, RTIL and gas meet. In this way, no slow step, such as analyte diffusion and/or dissolution in an electrolyte, is involved in the operative mechanism of RTIL-PEDs. Their current responses are instead conditioned by the sole analyte diffusion in gas phase to the PED surface, which is much faster than diffusion in liquid phase and it is just this type of rate limiting step that makes RTIL-PEDs fast-responding devices.
4. Conclusions
The results obtained in this investigation indicate that electrochemical detectors based on paper supported RTILs are very promising as electroanalytical sensors for the detection of gaseous analytes, not only because they allow electrochemical measurements to be performed in a medium where they are usually precluded or somehow difficult to be conducted. In fact, they make it possible to put together the advantages offered by the high electrical conductivity and negligible vapour pressure of RTILs and the benefits arising from their easy immobilization onto a porous and inexpensive supporting material such as paper for assembling membrane-free amperometric devices by resorting to a quite simple procedure.Moreover, by profiting from both a suitable dosage of the RTIL wicked on the paper and a careful screen printing of electrodes, it is possible to achieve an intimate contact between RTIL and electrode material at the probe surface so as to allow analytes to undergo charge transfer as soon as they reach the resulting interphase. This avoids the involvement of fairly slow steps such as analyte diffusion or dissolution in a conductive medium. In such a way, highly sensitive and fast-responding membrane-free PEDs can be assembled, which appear to be suitable for general applications.
Acknowledgements
Financial support from the Italian Ministry of University and Scientific Research is gratefully acknowledged.References
- P. A. Lieberzeit and F. L. Dickert, Anal. Bioanal. Chem., 2007, 387, 237–247 CAS.
- L. I. B. Silva, A. C. Freitas, T. A. P. Rocha-Santos, M. E. Pereira and A. C. Duarte, Talanta, 2011, 83, 1586–1594 CrossRef CAS.
- A. Berna, Sensors, 2010, 10, 3882–3910 CrossRef CAS.
- G. Bontempelli, N. Comisso, R. Toniolo and G. Schiavon, Electroanalysis, 1997, 9, 433–443 CAS.
- R. Knake, P. Jaquinot, A. W. E. Hodgson and P. C. Hauser, Anal. Chim. Acta, 2005, 549, 1–9 CrossRef CAS.
- G. Schiavon, G. Zotti and G. Bontempelli, Anal. Chim. Acta, 1989, 221, 27–41 CrossRef CAS.
- L. Loub, F. Opekar, V. Pacakova and K. Stulik, Electroanalysis, 1992, 4, 447–451 CAS.
- R. Toniolo, G. Bontempelli, G. Schiavon and G. Zotti, J. Electroanal. Chem., 1993, 356, 67–80 CrossRef CAS.
- L. R. Jordan and P. C. Hauser, Anal. Chem., 1997, 69, 2669–2672 CrossRef CAS.
- F. Opekar and K. Stulik, Anal. Chim. Acta, 1999, 385, 151–152 CrossRef CAS.
- R. Toniolo, P. Geatti, G. Bontempelli and G. Schiavon, J. Electroanal. Chem., 2001, 514, 123–128 CrossRef CAS.
- R. Toniolo, N. Comisso, G. Schiavon and G. Bontempelli, Anal. Chem., 2004, 76, 2133–2137 CrossRef CAS.
- J. Gebicki and B. Chachulski, Electroanalysis, 2009, 21, 1568–1576 CrossRef CAS.
- R. Toniolo, S. Susmel, N. Dossi, A. Pizzariello, M. Martinis and G. Bontempelli, Electroanalysis, 2010, 22, 645–652 CrossRef CAS.
- M. C. Buzzeo, C. Hardacre and R. G. Compton, Anal. Chem., 2004, 76, 4583–4588 CrossRef CAS.
- R. Toniolo, A. Pizzariello, S. Susmel, N. Dossi, A. P. Doherty and G. Bontempelli, Electroanalysis, 2007, 19, 2141–2148 CrossRef CAS.
- X. J. Huang, L. Aldous, A. M. O'Mahony, F. Javier del Campo and R. G. Compton, Anal. Chem., 2010, 82, 5238–5245 CrossRef CAS.
- Z. Silver and R. Bookman, Anal. Chem., 1956, 28, 558 CrossRef.
- J. Sherma and B. Fried, Anal. Chem., 1984, 56, 48R–63R CAS.
- D. A. Bruzewicz, M. Reches and G. M. Whitesides, Anal. Chem., 2008, 80, 3387–3392 CrossRef CAS.
- A. W. Martinez, S. T. Phillips, B. J. Wiley, M. Gupta and G. M. Whitesides, Lab Chip, 2008, 8, 2146–2150 RSC.
- E. Carrilho, A. W. Martinez and G. M. Whitesides, Anal. Chem., 2009, 81, 7091–7095 CrossRef CAS.
- W. Dungchai, O. Chailapakul and C. S. Henry, Analyst, 2011, 136, 77–82 RSC.
- A. K. Ellerbee, S. T. Phillips, A. C. Siegel, K. A. Mirica, A. W. Martinez, P. Striehl, N. Jain, M. Prentiss and G. M. Whitesides, Anal. Chem., 2009, 81, 8447–8452 CrossRef CAS.
- J. L. Delaney, C. F. Hogan, J. Tian and W. Shen, Anal. Chem., 2011, 83, 1300–1306 CrossRef CAS.
- J. Yu, L. Ge, J. Huang, S. Wang and S. Ge, Lab Chip, 2011, 11, 1286–1291 RSC.
- W. Dungchai, O. Chailapakul and C. S. Henry, Anal. Chem., 2009, 81, 5821–5825 CrossRef CAS.
- Z. Nie, C. A. Nijhuis, J. Gong, X. Chen, A. Kumachev, A. W. Martinez, M. Narovlyansky and G. M. Whitesides, Lab Chip, 2010, 10, 477–483 RSC.
- Z. Nie, F. Deiss, X. Liu, O. Akbulut and G. M. Whitesides, Lab Chip, 2010, 10, 3163–3169 RSC.
- M. K. Halbert and R. P. Baldwin, Anal. Chem., 1985, 57, 591–595 CrossRef CAS.
- E. Khaled, G. G. Mohamed and T. Awad, Sens. Actuators, B, 2008, 135, 74–80 CrossRef.
- A. Napier and J. P. Hart, Electroanalysis, 1996, 8, 1006–1013 CAS.
- L. E. Barrosse-Antle, A. M. Bond, R. G. Compton, A. M. O'Mahony, E. I. Rogers and D. Silvester, Chem.–Asian J., 2010, 5, 202–230 CrossRef CAS.
- A. P. dos Rios, F. J. Hernandez-Fernandez, F. Tomas-Alonso, J. M. Palacios, D. Gomez, M. Rubio and G. Villora, J. Membr. Sci., 2007, 300, 88–94 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |