DOI:
10.1039/C1LC20619B
(Paper)
Lab Chip, 2012,
12, 159-166
Meniscus induced self organization of multiple deep concave wells in a microchannel for embryoid bodies generation†
Received
11th July 2011
, Accepted 4th October 2011
First published on 10th November 2011
Abstract
Embryonic stem cells (ESCs) have attracted great interest in the fields of tissue engineering, regenerative medicine, and organogenesis for their pluripotency and ability to self-renew. ESC aggregation, which produces an embryoid body (EB), has been widely utilized as a trigger of in vitro directed differentiation. In this paper, we propose a novel method for constructing large numbers of deep concave wells in PDMS microfluidic chips using the meniscus induced by the surface tension of a liquid PDMS prepolymer, and applied this chip for the mass production of uniform sized EBs. To investigate if the microenvironment in the deep concave well is suitable for ES cells, the oxygen diffusion to the deep concave well was analyzed by CFD simulation. Murine EBs were successfully formed in the deep concave wells without loss of cells and laborious careful intervention to refresh culture media. The size of the EBs was uniform, and retrieving of EBs was done just by flipping over the chip. All the processes including EB formation and harvest are easy and safe to cells, and their viability after completion of all processes was over 95%. The basic properties of the EBs were generated and their capacity to differentiate into 3 germ layers was investigated by analyzing the gene expression profile. The harvested EBs were found to differentiate into cardiac cells and neurons, and neurofilaments formed branches of elongated extensions more than 1.0 mm in length.
Introduction
Embryonic stem cells (ESCs) are pluripotent, self-renewable, and can differentiate into more than 200 cell types in the adult body.1–4 These properties of ESCs have attracted much attention in the fields of regenerative medicine,5,6 tissue replacement after injury or disease7,8 and bio-pharmaceuticals.8,9 The formation of embryoid bodies (EBs) by ESCs has been utilized widely as a trigger for in vitro differentiation. EBs can generate cells of all germ layers upon treatment with appropriate cytokines, growth factors, and vitamins. A critical issue in ESC research is the inability to direct homogeneous differentiation into specific lineages, and control over the microenvironment to produce monodisperse EBs is very important for addressing this issue.10–12 Unfortunately, conventional methods, including scraping techniques,13 hanging drops, and suspension culture methods,14,15 have not lent themselves to the resolution of such problems. Recent progress in microtechnology has been widely applied to ES cell research.16,17 Especially, the microfabrication technology has led to the development of various microscale arrayed wells to promote uniform EB formation. Non-adhesive polyethylene glycol (PEG) microwell arrays18 and polyurethane microwells containing self-assembled monolayers19,20 have been widely used to control the homogeneity of EB size and shape. Microcontact-printed substrates (200–800 μm in diameter) have been used to regulate EB-size-dependent ES cell differentiation and to explore the underlying stem cell biology.21,22 Although these methods demonstrated the successful formation of EBs, some limitations have remained. The contour of cylindrical microstructures formed by the PEG microwells do not follow the curvature of EBs, and microcontact-printed substrates can only control the initial size of EBs.11,18 Furthermore, the loss of cells during the seeding process, uneven seeding densities, and difficulties in providing signaling chemicals and in changing the culture media are other disadvantages of these methods. Some researchers have developed microfluidic chips for EB formation to overcome these shortcomings, but obtaining large uniform populations of EBs, minimizing cell loss, and facilitating their harvesting from the chips are still remaining challenges.23 For the minimization of cell loss during EB formation, the pyramidal microwell arrays were proposed by Ungrin et al.24 We previously described the use of a concave PDMS microwell array with wells that were similar in contour to that of EBs. We discovered that EBs formed well in a controlled homogeneous manner in the concave microstructures, and they were retrieved easily after culturing for 4 days in vitro.25 Yet, still, two significant challenges have hindered the widespread use of these microwell arrays. In addition, the ESCs in the open arrayed wells are cultured in petri dishes, and culture media must be changed carefully to prevent escape of the grown EBs. Such media change is laborious work and it is preferable that this step be minimized. The integration of microwells in the microfluidic channel has been proposed as a possible solution to overcome this problem.
In this paper, we propose a novel method for constructing a large number of deep concave wells in PDMS microfluidic chips using the meniscus induced by the surface tension of a liquid PDMS prepolymer. To investigate whether the microenvironment in the deep concave well is suitable for ES cells, the oxygen diffusion to the deep concave well was analyzed by CFD simulation. Murine EBs were successfully formed in the deep concave wells without loss of cells and laborious careful intervention to refresh culture media. Their sizes were uniform, and the loss of ESCs was minimized by the appropriate design of the well shape. The EBs were easily harvested by flipping over the chip. The basic properties of the EBs were evaluated and their capacity to differentiate into 3 germ layers were investigated by analyzing the gene expression profile for 12 days. The harvested EBs were found to differentiate into cardiac cells and neurons, and neurofilaments formed branches of elongated extensions more than 1.0 mm in length.
Materials & methods
Preparation of PDMS channel integrating deep concave wells
Concave microwells were fabricated by taking advantage of the meniscus induced by the surface tension of a PDMS prepolymer. Initially, a PDMS microfluidic channel (base channel) was constructed using soft lithography by the replication of an SU-8 (MicroChem, MA, USA) master mold. The base channel was composed of pentagonal chambers in which the concave wells could be constructed. The top of the base channel was covered with a PDMS membrane (thickness ≈ 1 mm) to squeeze the PDMS prepolymer in the pentagonal chambers. To fabricate concave microwell chambers, a PDMS prepolymer (mixture of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 silicon elastomer (Sylgard 184) and curing agent) was introduced into the base channel (Fig. 1a) until it filled all pentagonal chambers. By pressing and rolling (P&R) the soft top of the base channel with a cylindrical rod 3–5 times, the prepolymer in the pentagonal chamber was extruded to the outlet, and the extruded prepolymer was removed by suction (Fig. 1b, left). The P&R process was crucial to the formation of concave structures. If the prepolymer were removed only by the suction force from the outlet, the prepolymer in the pentagonal chambers could not be removed. A detailed diagram showing the effect of the P&R process is shown in Fig. 1b (right). After squeezing, the chip was vertically positioned, and the prepolymer remaining in the pentagonal chamber generated a meniscus inside the pentagonal well due to the surface tension and the bottom of the pentagonal chamber became a concave structure (Fig. 1c). The self-organized concave structures were thermally cured (80 °C for 1 h), and a large number of concave wells were constructed inside the microfluidic channel (Fig. 1d). The size and depth of each well were determined by the shape of the pentagonal chambers. Three types of pentagonal chambers, with widths of 300, 500, or 700 μm, were used to form multi-sized concave wells.
:1 silicon elastomer (Sylgard 184) and curing agent) was introduced into the base channel (Fig. 1a) until it filled all pentagonal chambers. By pressing and rolling (P&R) the soft top of the base channel with a cylindrical rod 3–5 times, the prepolymer in the pentagonal chamber was extruded to the outlet, and the extruded prepolymer was removed by suction (Fig. 1b, left). The P&R process was crucial to the formation of concave structures. If the prepolymer were removed only by the suction force from the outlet, the prepolymer in the pentagonal chambers could not be removed. A detailed diagram showing the effect of the P&R process is shown in Fig. 1b (right). After squeezing, the chip was vertically positioned, and the prepolymer remaining in the pentagonal chamber generated a meniscus inside the pentagonal well due to the surface tension and the bottom of the pentagonal chamber became a concave structure (Fig. 1c). The self-organized concave structures were thermally cured (80 °C for 1 h), and a large number of concave wells were constructed inside the microfluidic channel (Fig. 1d). The size and depth of each well were determined by the shape of the pentagonal chambers. Three types of pentagonal chambers, with widths of 300, 500, or 700 μm, were used to form multi-sized concave wells.
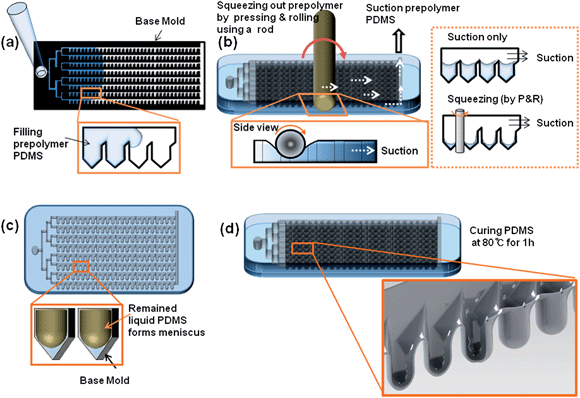 |
| | Fig. 1 Schematic diagram of the fabrication of an array of meniscus-induced deep concave wells in a microfluidic chip; (a) PDMS prepolymer was introduced into the base channel of the microfluidic chip, (b) PDMS prepolymer in the microfluidic chip was squeezed out by pressing and rolling using a rod, (c) liquid PDMS remaining in the microfluidic chip spontaneously formed a meniscus, and finally (d) a deep concave microwell array was fabricated using the meniscus template. | |
Murine ES cell preparation and seeding
The R1 ES cell line (American Type Culture Collection, Manassas, VA) used in this experiment was a sub-cell line of J1 ES cells established by Dr Rudolph Jaenisch (Whitehead Institute for Biomedical Research, Cambridge, MA). Cells were cultured on gelatin-coated dishes with medium consisting of 15% ES-qualified fetal bovine serum (FBS, Invitrogen, CA) and 1400 Unit ml−1leukemia inhibitory factor (LIF, Millipore, MA) in Dulbecco's Modified Eagle Medium (DMEM, Invitrogen, CA) Knockout medium. Cells were passaged every 3 days by dissociating the cells into small colonies using 0.25% trypsin (Invitrogen, CA) and replating on a 0.1% gelatin-coated dish at a subculture ratio of 1:6. To seed the cells in the concave wells, cell suspensions were prepared with densities of 6.0, 9.0, and 12.0 × 106cells ml−1 and were introduced to the chip through the inlet port (Fig. 2a). Two-hundred microlitres of the ES cell suspension at each density were injected into the microfluidic channel through the inlet port. After filling the channel, the microfluidic chip was maintained in a 37 °C incubator in a vertical position for 30 min to allow the cells to be trapped in the concave microwells (Fig. 2b). A schematic depiction of loss-free cell aggregation in the concave microwells is illustrated in Fig. 2b (right). By slanting the top of the chamber walls, all cells became trapped at the bottom of the concave microwells due to the gravity.
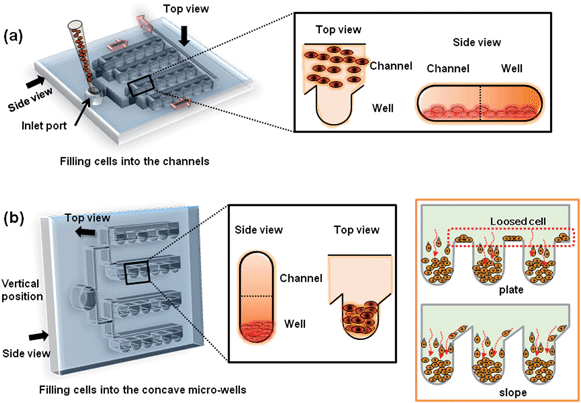 |
| | Fig. 2 Schematic diagram of the process of cell seeding in the deep concave microwells arrayed on the microfluidic chip; (a) an mESC suspension was introduced to the inlet port of the microfluidic chip through the microfluidic channels (red arrows indicate the direction of flow). At this step, the cells were positioned at the bottom of each microfluidic channel. A top and side view of a microfluidic channel and well are shown in the black box. (b) To trap mESCs into microwells via gravity, the microfluidic chip was vertically positioned in an incubator (black box). The slope in the microwell minimized the cell loss. Furthermore, no washing processes were required to remove loosened cells (orange box). | |
Computational fluid dynamics analysis
The chips were designed with consideration for the oxygen distribution in the deep concave wells, which is an important factor for cell viability. Prior to designing and fabricating the microfluidic chips, we performed a two-dimensional time-dependent simulation of oxygen diffusion in a microfluidic chip using a finite element method (FEM)-based computational fluid dynamics (CFD) code in the COMSOL Multiphysics 4.0 software package (COMSOL Inc., MA, USA). A triangular mesh system was generated to model the microchannels and microwells, and the number of grids was 17391. The diffusion coefficient of oxygen in the medium was assumed to be the same as that in water (3.1 × 10−9 m2 s−1). The initial concentration of oxygen in the medium in a microchannel was assumed to be 0.119 mol m−3, and in the microwells the initial concentration of oxygen was assumed to be zero. The diffusion of oxygen was estimated based on Fick's second law 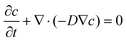 , in which c denotes solute concentration (mol m−3), and D is the diffusion coefficient (m2 s−1) of the solute. We set all boundaries to no-mass flow-through.
, in which c denotes solute concentration (mol m−3), and D is the diffusion coefficient (m2 s−1) of the solute. We set all boundaries to no-mass flow-through.
Cell viability test
Cell viability tests were performed by adding 5 mL phosphate-buffered saline (PBS) containing 2 μL of calcein AM solution and 10 μL ethidium homodimer-1 solution to the microchannels, followed by incubation at 37 °C in a 5% CO2 incubator for 40 min. The stained ES cells were analyzed using an inverted fluorescence microscope (EVOS, AMG, USA). Calcein AM fluoresces in green to permit detection of live cells, whereas the ethidium homodimer-1 fluoresces in red to indicate dead cells. The survival rate of the ES cells was measured by image analysis using Image J (http://rsbweb.nih.gov/ij/).
Three germ layer differentiation of EBs and gene analysis
Murine ES cells were seeded and cultured within each of the three sizes of concave microwells for 3 days in vitro. The EBs formed within concave microwells were gently retrieved and transferred in Knockout-DMEM containing 15% FBS, non-essential amino acids, 2 mM L-glutamine, mercaptoethanol (Invitrogen, CA), and 1% penicillin-streptomycin. After the EBs had been cultured for 3, 6, 9, and 12 days in vitro, they were digested in TRIzol Reagent (Invitrogen, CA), followed by chloroform extraction and precipitation with isopropyl alcohol. cDNA was synthesized from the purified RNA using reverse transcriptase (TAKARA, Japan) as per the manufacturer's instructions.
Culture conditions for ES cell-derived cardiac and neuronal differentiation and immunostaining analysis
EBs cultured in concave microwells for 3 days in vitro were retrieved and transferred to tissue culture dishes. The differentiation study to cardiac and neuron cells were performed to confirm the differentiability of EBs to diverse cell lineage. After 3 days, EBs were cultured with cardiac differentiation culture medium, MEM-alpha (Gibco), 15% FBS, 1% penicillin/streptomycin, and with neuronal differentiation medium supplemented with insulin/transferrin/selenium/fibronectin (ITSFn) medium (Invitrogen, CA) respectively. The culturing medium was replaced every other day. The cardiac beating and the neuroepithelial progenitor cells from the EBs were observed after culturing for 8 days. The lengths of the neurofilament branches were quantified using Image J (http://rsbweb.nih.gov/ij/). The cells cultured for 11 days were fixed for 20 min with 4% formaldehyde at 4 °C. Cells were permeabilized by using 0.1% Triton-X100 in 0.1% PBS for 20 min at room temperature, were blocked with 3% BSA in PBS for 30 min, and then were incubated with primary antibody overnight at 4 °C. Primary antibodies (Stemcell Technologies, Canada) against neurofilament (1:1000), nestin (1:100), and sarcomeric α-actinin (1:100) were used to characterize various cell types. After incubating overnight, cells were washed with PBSA (0.1% BSA in PBS) for 5 min. Secondary antibodies (1:1000 dilutions, Invitrogen, CA) were applied for 1.5 h at room temperature. Cells were again washed with PBSA, and fluorescent images were acquired by using a fluorescence microscope (EVOS, AMG, USA) after counterstaining with 4,6-diamidino-2-phenylindole dihydrochloride (DAPI, Invitrogen, CA).
Results
Concave microwell-array in microfluidic chip
Fig. 3(a) illustrates the fabricated EB-forming chip with a total of 300 concave wells (width: 700 μm). The size of the chip was 20 mm × 40 mm, and the thickness was 5 mm, comparable to that of a slide glass. The inset shows that the microwells were fabricated homogeneously with a concave bottom structure. Throughout all types of chip (widths of the pentagonal chambers: 300, 500, and 700 μm), concave structures were successfully self-organized by the P&R process only. The SEM image shows the smooth bottom profile of the concave wells.
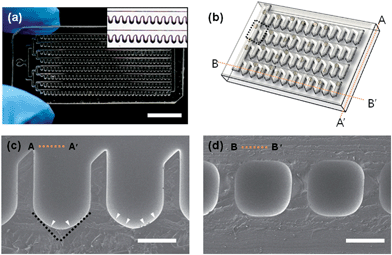 |
| | Fig. 3 A meniscus-induced deep concave microwell array microfluidic chip; (a) optical microscopy image of the microfluidic chip (the scale bar indicates 1 mm), the inset shows magnified images of the deep concave microwell array. (b) Schematic of the concave microwell arrayed microfluidic chip, lines AA′ and BB′ indicate the cross section line of (c) and (d), respectively. SEM images of the cross section view of line AA′ (c) and line BB′ (d) show the bottom of a microwell, the black dotted line indicates the edge of the pentagonal microwell, and the white arrowhead indicates the rounded shape induced by the meniscus (scale bar: 500 μm). | |
Computational analysis of oxygen diffusion
Fig. 4 presents the computational results of oxygen diffusion in a concave well. We assumed that the initial oxygen concentration in a concave well is zero, and oxygen diffuses from the main flow channel into a concave well. The simulation results illustrate that oxygen in the flow channel rapidly starts to diffuse into a concave well after 10 s (Fig. 4(a)). The quantitative oxygen concentration in a well is plotted as a function of time in Fig. 4(b). Within 60 s, approximately 70% of the equilibrium oxygen concentration had diffused through a well, and 95% oxygen diffusion was achieved within 200 s.
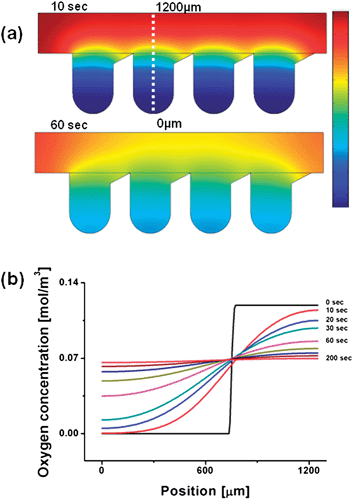 |
| | Fig. 4 Computational prediction of the oxygen diffusion profile in the concave well array; (a) the simulation results indicated that oxygen in the flow channel would rapidly diffuse into the concave wells after 10 s, and oxygen was mainly distributed over all concave wells within 60 s. (b) The time-dependent quantitative oxygen concentration from a well bottom to a channel. Within 60 s, approximately, the oxygen levels reached 70% of the equilibrium value by diffusion through the well, and 95% oxygen levels were achieved within 200 s. | |
EB generation in the microfluidic chip
Murine ES cells were seeded in the microfluidic system by introducing 200 μl cell suspension medium through the inlet channel. When the cell suspension medium had filled the volume of the microchannel, we stopped the flow and positioned the chip vertically. The cells in the suspension medium then sank into the concave wells by gravity and were spontaneously trapped in the concave wells with minimum cell loss. Fig. 5(a) illustrates ES cells in the concave wells after seeding. The number of cells trapped in each concave well was narrowly distributed. One day after seeding, ES cells self-aggregated and formed round shapes on the concave surface of each well (Fig. 5(b)), and EBs were stably formed at day 3. The Live/Dead test results at day 3 showed that more than 95% EBs were viable (Fig. 5(c)). EBs were harvested from the concave wells after culturing for 3 days. By flipping the chip upside down and tapping gently with fingers for a few seconds (5–10 times), most of the EBs separated from the concave wells and were extruded with the media flowing through the main channel. Fig. 5(d) illustrates the detached EBs flowing through the channel, and the harvesting rate exceeded 98%. Fig. 5(e) shows the homogenous sizes of the harvested EBs. The inset of Fig. 5(e) demonstrates that a large number of EBs were generated on the microfluidic chip.
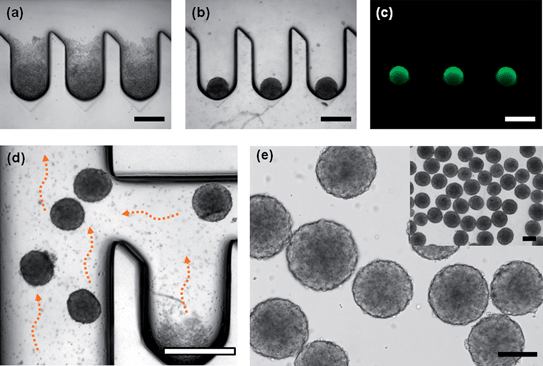 |
| | Fig. 5 EBs formation in the deep concave microwell arrayed microfluidic chip; (a) Murine ES cells were seeded in the microfluidic system (day 1). After seeding, uniform numbers of cells were trapped in each concave well. (b) ES cells self-aggregated in a spherical shape on the concave surface of a well (day 2). (c) EBs were stably formed, and EBs were removed from the microwells by gentle tapping after culturing for 3 days. (d) EBs were harvested from the microfluidic chip by introducing culture medium. (e) The homogenous size of the harvested EBs. The inset shows a large number of EBs generated in a single microfluidic chip (all scale bars indicate 400 μm). | |
Evaluation of harvested EBs and differentiation to cardiac and neuronal cells
The viability of the harvested EBs was tested, and Fig. 6(a) shows the EBs stained with the Live/Dead test kit. Most of the fluorescent images of the EBs exhibited green fluorescence, indicating excellent viability. The survival rate of the harvested EBs was 95%, similar to the viability of EBs grown in the concave wells. This indicates that the EBs were not damaged during the harvesting process. The harvested EBs were monodisperse and the breadth of the size distribution was less than 15% for most EBs formed in the 300, 500, and 700 μm wells. The size of the EBs could be controlled according to the well size and cell density (Fig. 6(b)). The size of the EBs was sensitive to the cell density (6.0 × 106, 9.0 × 106, and 1.2 × 107cells ml−1), even if the EBs were formed in concave wells of the same size. The EB size was linearly correlated with the cell density, particularly in the 300 and 500 μm concave wells. The EBs were harvested from 500 μm concave wells, transferred to tissue culture dishes, and cultured in the differentiation medium containing MEM-alpha and ITSFn (20 mg mL−1) for an additional 8 days, respectively, resulting in cardiac (Fig. 6c) and neuroepithelial differentiation (Fig. 6d). To analyze cardiogenesis of the EBs, the differentiated cells were immunostained with sarcomeric α-actinin and DAPI for cardiac differentiation and neurofilament, nestin, and DAPI for neuronal differentiation. The neurofilament formed branches of elongated extensions more than 1.0 mm in lengths, which demonstrate the potential to generate neurite outgrowth.
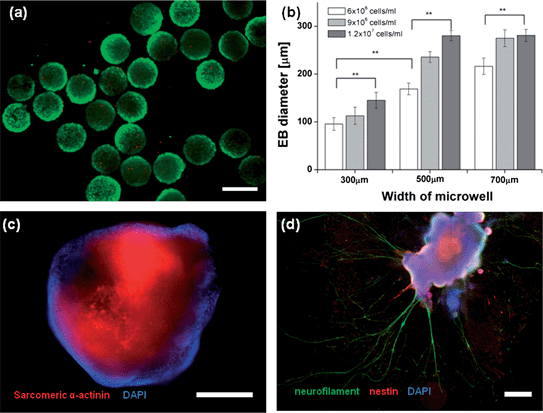 |
| | Fig. 6 Evaluation of the harvested EBs and neuronal differentiation; (a) the viability of harvested EBs was tested. Most of the fluorescent images of EBs exhibited green fluorescence (live), indicating excellent viability. The survival rate of the harvested EBs exceeded 95% (the scale bar indicates 300 μm). (b) The harvested EBs were monodisperse, and the width of the size distribution was less than 10% for most EBs formed in the 300, 500, and 700 μm wells. The size of EBs could be tuned according to the well size and cell density. Error bars indicate the standard deviation; ** indicate p < 0.01 compared to 300 μm EBs (n = 20; Student's t-test). (c) The fluorescent image shows cardiac differentiation of an EB (scale bar: 400 μm, the figure was generated from 2 fluorescent images (10×)). (d) The figure illustrates neuronal differentiation of an EB. The length of the neurite exceeded 1 mm (scale bar: 400 μm, the figure was generated from 35 fluorescent images (10×)). | |
We analyzed three germ layer differentiation of EBs cultured in concave microwells by evaluating the expression of certain gene markers on day 3, 6, 9 and 12 (Fig. 7a). The analysis was performed at a high cell density (12.0 × 106cells ml−1). We found no evidence for the expression of the endodermal differentiation marker, AFP, in any of the microwells on day 3. On day 9, however, AFP was strongly expressed in EBs grown in all microwells (300, 500, and 700 μm in width), indicating that endodermal differentiation was mainly enhanced by the culturing period. An analysis of the time-dependent expression of the marker for ectoderm differentiation, nestin, showed that the expression of ectoderm was not significantly different among the microwells at day 3 to 12. Nestin expression also depended on the culturing time and was increased with elapse of time. Mesoderm differentiation based on expression of the bone morphogenetic protein 4 (BMP-4) marker did not display a significant size dependence for culturing days. This analysis also shows that cells differentiated into mesoderm and ectoderm earlier than the endoderm differentiation. The endoderm was formed later than other germ layers.
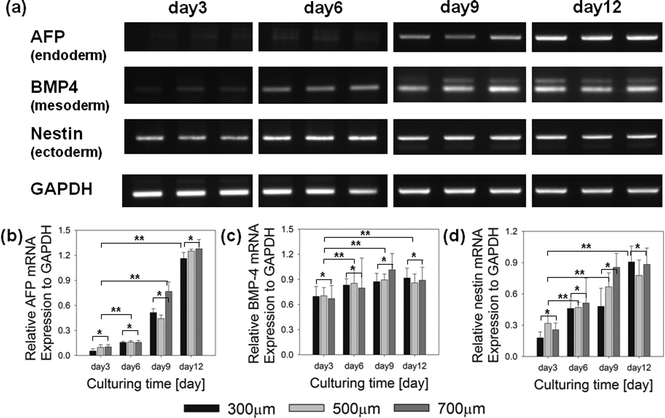 |
| | Fig. 7 Time-course of the expression of markers for the three germ layers in EBs. (a) Representative figures showing the expression of mRNA for the three germ layer markers in the ES cells cultured within concave microwells for day 3 to day 12 in vitro. (b) Quantification of the relative gene expression of AFP (endoderm), (c) BMP-4 (mesoderm), and (d) nestin (ectoderm) on day 3 to 12. The AFP, endoderm marker, strongly expressed after culturing day 9. The gene expression results of nestin, mesoderm marker, gradually increase with elapse of culturing day. The error bars indicate the standard deviation, *p < 0.05, and **p < 0.01, respectively, compared to 300 μm EBs (n = 4; Student's t-test). | |
Discussion
A microfluidic chip that included an array of deep concave microwells for EB production was successfully fabricated by taking advantage of the meniscus formed by squeezing of the PDMS prepolymer deposited in pentagonal microchambers. In general, fabrication of curved structures, including concave or convex shapes using conventional micro-fabrication techniques, is challenging and requires skill, facilities, and complicated processes. Although several simple and cost-effective methods exist for fabricating microwell arrays, including ice droplet-based microcavity formation,26 cylindrical microchannel formation using a water mold,27 replication of elastic membrane deflection25,28 and meniscus effects,29,30 it is challenging to integrate deep concave structures in PDMS microfluidic channels without the use of complicated processes. The meniscus-based-process proposed here overcomes these limitations using an extremely simple and easy method. Following preparation of a base channel that includes pentagon chambers using conventional soft-lithography techniques, the fabrication of deep concave wells is straightforward, and the fabrication time is 2 h. Labor, experienced skill, devices, and facilities are not required at all. This technique is anticipated to be widely applicable to the fabrication of more complicated round structures to achieve further diverse applications in biomedical research.
The unique feature of our method is the squeezing of the PDMS prepolymer. The CFD simulation study demonstrated that using only the suction applied to the outlet may remove the prepolymer in the main channel. However, in the pentagonal chamber, the flow rate induced by the suction force was approximately zero and the prepolymer could not be eliminated (Fig. S2a, ESI†). Thus, construction of deep concave wells is almost impossible using this method, and all pentagonal chambers were filled with the PDMS prepolymer as illustrated in Fig S2(b).† In contrast, squeezing of the introduced prepolymer by pressing and rolling with a rod removed the prepolymer in pentagonal chambers to some extent, thereby forming a concave surface at the bottom of the pentagon chambers.
In employing the chip for the preparation of uniformly sized EBs, it is critical to seed the same number of cells to all wells and to ensure that oxygen diffuses into the deep concave wells. The CFD-based simulation study predicted that the oxygen content reached almost 70% of its maximum value by diffusion within 60 s, and the oxygen concentration in the concave well approached that of freshly provided media within 210 s. This result shows that oxygen was easily delivered to cellsvia diffusion, even those cells located at the bottom of the deep concave wells. A similar numbers of ES cells were trapped in each concave well to form uniformly sized EBs. Due to the slope of the top of the well and to proper control over the flow rate, as shown in Fig. 2, uniform cell trapping was achieved without loss of cells, which is another crucial feature of the proposed chip.
During differentiation of the ES cells, the size of the EB affects the differentiation characteristics, and precise control over the EB size is significant.11,25 Usually, the size of an EB can be controlled by changing the diameter of the concave well. Interestingly, we showed that the size could also be controlled by changing the cell density. The diameter of EBs varied almost linearly with the cell density. This property suggests that the microfluidic chip developed in this study may be a potentially useful tool for achieving size-sensitive differentiation of ES cellsin vitro. The differentiation capability of the EBs into 3 germ layers was evaluated by the expression of gene markers on day 3, 6, 9, and 12, and three representative markers, AFP (endoderm), nestin (ectoderm), and BMP-4 (mesoderm), were used to analyze the germ layer differentiation capabilities (http://stemcells.nih.gov/info/scireport/appendixe.asp%23eii). As reported by several researchers, including us, differentiation was critically affected by the culturing time.25,31,32 The ectoderm and mesoderm genes, nestin and BMP-4, showed stronger expression than AFP at both days 3 and 6. However, the endoderm marker strongly appeared at day 9 and 12. These results show that the stem cells differentiated into ectoderm and mesoderm earlier than their differentiation into endoderm in concave microwells. The mesoderm gradually differentiated in concave microwells as time elapsed. These gene studies agree with previously reported results,21 suggesting that the production of EBs using the chip provided EBs with characteristics that were identical to those of EBs generated by other methods. The long neurofilament formation over 1.0 mm in length demonstrated the capability of EBs to differentiate into specific cell lineage. Another significant result of this study is that EB formation was fully performed inside the microfluidic chip just after seeding the cell suspension media without direct intervention. This suggests that automation of the EB production process and sequential application of diverse biochemicals during EB formation are feasible using the proposed chip. This feature involves the development of novel tool for controlling the differentiation process and for enhancing stem cell research.
Conclusion
We developed a concave microwell arrayed microfluidic chip that can be fabricated inexpensively using minimal labor by taking advantage of a simple meniscus effect. Our method shows that complicated micro-round structures could be easily fabricated. The microfluidic chip enabled size control over EB formation and showed that EBs of a controlled size could be generated on a single chip with minimal cell loss. The EBs could be harvested from the microchannels without damage simply by tapping on the reverse side of the substrate. The EBs maintained their capacity to differentiate into 3 germ layers, and we observed the neural differentiation of the generated EBs.
Acknowledgements
This paper is supported by the Korea Science and Engineer Foundation (KOSEF) (grant no. 20110020455) and this work was also supported by the Korea Research Foundation Grant funded by the Korean Government (MEST) (KRF-2008-220-D00133).
References
- B. E. Reubinoff, M. F. Pera, C. Y. Fong, A. Trounson and A. Bongso, Nat. Biotechnol., 2000, 18, 399–404 CrossRef CAS.
- J. S. Odorico, D. S. Kaufman and J. A. Thomson, Stem Cells, 2001, 19, 193–204 CrossRef CAS.
- V. F. M. Segers and R. T. Lee, Nature, 2008, 451, 937–942 CrossRef CAS.
- B. Bhattacharya, S. Puri and R. K. Puri, Curr. Stem Cell Res. Ther., 2009, 4, 98–106 CrossRef CAS.
- Y. Xu, Y. Shi and S. Ding, Nature, 2008, 453, 338–344 CrossRef CAS.
- N. S. Hwang, S. Varghese, H. J. Lee, Z. Zhang, Z. Ye, J. Bae, L. Cheng and J. Elisseeff, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 20641 CrossRef CAS.
- O. Caspi, A. Lesman, Y. Basevitch, A. Gepstein, G. Arbel, M. Habib, L. Gepstein and S. Levenberg, Circ. Res., 2007, 100, 263–272 CrossRef CAS.
- G. Q. Daley and D. T. Scadden, Cell, 2008, 132, 544–548 CrossRef CAS.
- F. Mannello and G. A. Tonti, Stem Cells, 2007, 25, 1603–1609 CrossRef CAS.
- Y. Sakai and K. Nakazawa, Acta Biomater., 2007, 3, 1033–1040 CrossRef CAS.
- Y. S. Hwang, B. G. Chung, D. Ortmann, N. Hattori, H. C. Moeller and A. Khademhosseini, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 16978 CrossRef CAS.
- D. Nguyen, S. Sa, J. D. Pegan, B. Rich, G. Xiang, K. E. McCloskey, J. O. Manilay and M. Khine, Lab Chip, 2009, 9, 3338–3344 RSC.
- J. Itskovitz-Eldor, M. Schuldiner, D. Karsenti, A. Eden, O. Yanuka, M. Amit, H. Soreq and N. Benvenisty, Molecular Medicin, 2000, 6, 88–95 CAS.
- G. M. Keller, Curr. Opin. Cell Biol., 1995, 7, 862–869 CrossRef CAS.
- A. Shiroi, M. Yoshikawa, H. Yokota, H. Fukui, S. Ishizaka, K. Tatsumi and Y. Takahashi, Stem Cells, 2002, 20, 284–292 CrossRef CAS.
- J. Y. Park, S. K. Kim, D. H. Woo, E. J. Lee, J. H. Kim and S. H. Lee, Stem Cells, 2009, 27, 2646–2654 CrossRef CAS.
- J. Y. Park, S. Takayama and S. H. Lee, Integr. Biol., 2010, 2, 229–240 RSC.
- H. C. Moeller, M. K. Mian, S. Shrivastava, B. G. Chung and A. Khademhosseini, Biomaterials, 2008, 29, 752–763 CrossRef CAS.
- J. C. Mohr, J. J. de Pablo and S. P. Palecek, Biomaterials, 2006, 27, 6032–6042 CrossRef CAS.
- J. C. Mohr, J. Zhang, S. M. Azarin, A. G. Soerens and J. J. de Pablo, Biomaterials, 2010, 31, 1885–1893 CrossRef CAS.
- R. Peerani, B. M. Rao, C. Bauwens, T. Yin, G. A. Wood, A. Nagy, E. Kumacheva and P. W. Zandstra, EMBO J., 2007, 26, 4744–4755 CrossRef CAS.
- L. H. Lee, R. Peerani, M. Ungrin, C. Joshi, E. Kumacheva and P. W. Zandstra, Stem Cell Res., 2009, 2, 155–162 CrossRef CAS.
- Y. Torisawa, B. Chueh, D. Huh, P. Ramamurthy, T. M. Roth, K. F. Barald and S. Takayama, Lab Chip, 2007, 7, 770–776 RSC.
- M. D. Ungrin, C. Joshi, A. Nica, C. Bauwens and P. W. Zandstra, PLoS One, 2008, 3, e1565 Search PubMed.
- Y. Y. Choi, B. G. Chung, D. H. Lee, A. Khademhosseini, J. H. Kim and S. H. Lee, Biomaterials, 2010, 31, 4296–4303 CrossRef CAS.
- J. Y. Park, C. M. Hwang and S. H. Lee, Biomed. Microdevices, 2009, 11, 129–133 CrossRef.
- S. Chao, R. Carlson and D. R. Meldrum, Lab Chip, 2007, 7, 641–643 RSC.
- J. Y. Park, D. H. Lee, E. J. Lee and S. H. Lee, Lab Chip, 2009, 9, 2043–2049 RSC.
- K. Lee, C. Kim, K. S. Shin, J. W. Lee, B. K. Ju, T. S. Kim, S. K. Lee and J. Y. Kang, J. Micromech. Microeng., 2007, 17, 1533 CrossRef CAS.
- S. H. Lee, H. N. K. Do Hyun Kang and K. Y. Suh, Lab Chip, 2010, 10, 3300–3306 RSC.
- A. Leahy, J. W. Xiong, F. Kuhnert and H. Stuhlmann, J. Exp. Zool., 1999, 284, 67–81 CrossRef CAS.
- R. Chinzei, Y. Tanaka, K. Shimizu Saito, Y. Hara, S. Kakinuma, M. Watanabe, K. Teramoto, S. Arii, K. Takase and C. Sato, Hepatology, 2002, 36, 22–29 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1lc20619b |
| ‡ G. S. Jeong, and Y. Jun equally contributed to this study. |
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here.