On-chip sample preparation by controlled release of antibodies for simple CD4 counting
Markus
Beck
*,
Silvia
Brockhuis
,
Niels
van der Velde
,
Christian
Breukers
,
Jan
Greve
and
Leon W. M. M.
Terstappen
Medical Cell Biophysics, MIRA Institute for Biomedical Technology and Technical Medicine, Faculty of Science and Technology, PO Box 217, 7500 AE, Enschede, The Netherlands. E-mail: M.Beck@utwente.nl
First published on 3rd November 2011
Abstract
We present a simple system for CD4 and CD8 counting for point-of-care HIV staging in low-resource settings. Automatic sample preparation is achieved through a dried reagent coating inside a thin (26 μm) counting chamber, allowing the delayed release of fluorochrome conjugated monoclonal antibodies after the filling of the chamber with whole blood by capillary flow. A custom-built image cytometer is used to capture fluorescence images representing more than 1 μl of blood. The thin layer of blood in combination with the large image area allows the use of whole blood from a finger prick without the need for dilution, lysis or cell enrichment. Automatic cell counting of CD4+ and CD8+ T-lymphocytes correlates well with results obtained by flow cytometry.
Introduction
The number of CD4+ T-lymphocytes per microlitre of blood (CD4 count) is used as a measure of the effectiveness of antiretroviral therapy (ART) of patients infected with the human immunodeficiency virus (HIV),1 despite an ongoing discussion on its predictive value.2–4 For infants infected with HIV a CD8+ T-lymphocyte count is also acquired since the ratio of CD4 and CD8 better reflects the disease status.5 In most parts of the world, flow cytometry is successfully applied to carry out these tests.6 Several dedicated instruments for the purpose of CD4/CD8 counting are available. Examples are the instruments FACSCount (Becton Dickinson), Guava Easy CD4 (Millipore), PointCare NOW (PointCare) or Cyflow Counter (Partec).7 However, many rural areas, for example in Sub-Saharan Africa, the region most affected by HIV, do not have access to CD4 counting as they often lack the necessary infrastructure to ship blood samples to central labs and get the result back to the patient. Accordingly, there have been large efforts over the last decade8 to miniaturize flow cytometers9–13 or to develop alternative portable tests that can be carried out with minimal training,7,14 for example LabNow,15PIMA (Alere),16 Daktari,17–19 Zyomyx.20 Earlier, we developed a single platform image cytometer21 that simplified the instrumentation and provided very promising results,22–24 but still required precision for the sample preparation, which frequently cannot be met in the field by less experienced operators. Automated sample preparation is possible (e.g. in the PointCare NOW instrument), but the ambient conditions to be expected during operation (high temperature, large temperature differences, humidity ranging from almost 0% to 100%) impose enormous challenges on the long-term stability of such a system.We have developed a test that does not require any precision during sample preparation since the sample volume is given by the dimensions of a microfluidic counting chamber containing the dried fluorescently labelled antibodies and the image area of the imaging system. For CD4 and CD8 counting, we use the combinations of CD3-APC/CD4-PerCP and CD3-APC/CD8-PerCP, respectively, in separate test chambers. These chambers are filled with whole blood by capillary flow and after an incubation time of 30 min, the fluorescence is imaged with a CCD camera. Automated image analysis provides the results within 5–20 s on a current “standard” PC.
Principle: controlled release of antibodies
Fig. 1 illustrates the principle of the test. A glass chamber, coated on one side with a thin reagent layer, is filled by capillary flow with whole blood from a finger prick. During the filling of the chip, which takes approximately 1–2 s, the reagents stay within the layer, so that the initial uniform lateral distribution of reagent is maintained.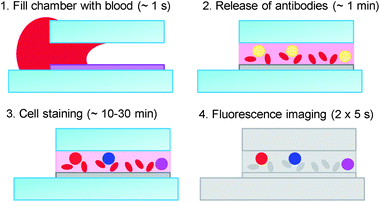 | ||
| Fig. 1 Principle of the test: blood from a finger prick fills the test chamber by capillary flow. During the filling process, which takes about one second, the antibodies stay in the hydrogel layer; on a time scale of several seconds to a few minutes, the layer swells and releases the antibodies into the blood; after the staining process (10–30 min), the fluorescence from the leukocytes is imaged and automated image analysis is used to determine the concentration of the cells of interest. | ||
In previous solutions, on-chip sample preparation in fluidic chips has typically been achieved either by mixing two fluids, which can be very challenging because turbulent flow is not easily realised in microfluidics,25 or through the binding of constituents of the sample (molecules, cells) to a functionalised surface, as in microarray technology.26 Hydrogels have been used to immobilize pH sensitive dyes,27antigens28 or antibodies29 and let the corresponding molecules contained in the sample diffuse into the gel. This approach, however, does not work for the staining of surface antigens on cells. Several sophisticated approaches for externally controlled release of reagent from reservoirs in a microfluidic chip have been reported.30–33 A solution using capillary delay valves34 and microchannels with different flow resistances (“reagent integrators”) for the passive release of reagent into a stream of sample has been described recently.35 In the PIMA Counter,16 a diagnostics instrument developed for simple CD4 counting, a lyophilised pellet containing the antibodies is actively mixed for 20 min with blood using a miniaturised peristaltic pump integrated into the cartridges. Controlled release of molecules from hydrogels has been extensively used for in vivo applications,36 to deliver drugs continuously over a long period (sustained release)37 or to delay the release (e.g. enteric coatings).38 These applications typically require release processes on time scales of many hours and therefore use layer thicknesses of typically 0.1–1 mm, whereas our application requires release times of about 10–100 s.
The release time of reagents can be tailored in several ways. First, different hydrogels can be used, similar to application-specific gels used for gel electrophoresis.39 Second, the thickness of the layer (the total amount of hydrogel) can be varied. Third, the deposition technique leaves room for optimization (e.g. process temperatures). The goal of this optimization is to ensure that the reagents are released quickly after the chip has been filled with blood, but not during the filling process, because this would result in an accumulation of reagent at the far end of the chip. Diffusion, especially of macromolecules such as antibodies, would not be sufficient to distribute the reagent uniformly across the layer during the incubation time. The diffusion constant of fluorochrome-conjugated antibodies in water at room temperature is on the order of 10 μm2 s−1. Even taking into account slightly reduced diffusion in blood serum, the antibodies will distribute uniformly across a chamber height of 25 μm within a few minutes, whereas the diffusion across the length of the chamber (12.5 mm) would take months.
Mixtures of gelatine and fluorochrome-conjugated monoclonal antibodies are cold-cast at room temperature to create reagent layers, which release the contained antibodies on the desired time scale of 10–100 s. Details of the fabrication procedure are given in the next section. Once the antibodies are released, cell staining with fluorescently labelled antibodies takes place as in any other immunofluorescent staining. To prevent drying of the blood during incubation, we seal the facets of the chip with mineral oil. In the future, this procedure will be replaced by self-sealing gelatine stripes at the openings of the chamber. As in a haemocytometer, the sample volume only depends on the height of the test chamber and on the imaged area. Since both parameters can be controlled precisely during the fabrication of instruments and fluidic chips, no precision is needed during sampling and instrument operation. Also, the low NA imaging allows for acceptable tolerances regarding the alignment between the test chamber and the camera, since the depth of focus is about 0.2 mm.
Typical immunofluorescent protocols use fluorochromes with different emission wavelengths to distinguish between cells labelled with different antibodies. However, since we use very low magnification imaging, even small chromatic aberration or misalignment related to the change of emission filters can result in image shifts exceeding the size of a cell, which can be difficult to compensate during image analysis. With the combination of the fluorochromes PerCP and APC, we eliminate the need to image two different emission wavelengths. This is possible because their emission spectra have a large overlap, with maxima in the far red spectral range at 670 nm and 685 nm, respectively, while their excitation spectra have only very little overlap with maxima at 480 nm and 630 nm, respectively. Therefore, PerCP and APC can be distinguished by taking two images through the same emission filter with blue and red excitation light, respectively. As described below, our excitation module has three colors, red, green and blue. A third fluorochrome could be used which is excited predominantly by green light (520–560 nm) with sufficient emission at λ > 650 nm. Examples are Nile Red, the tandem conjugates PE-Cy5, PE-Cy5.5, or the nucleic acid dye 7-AAD. The considerable overlap between the excitation spectra could be problematic, but compensation can be applied.
Experimental
Chip fabrication/reagent coating
The fluidic chambers are built from standard microscope slides (Menzel Gläser, Braunschweig, Germany), laminating adhesive (FL501, nominal thickness: 1 mil = 25.4 μm, 3 M, St Paul, MN, USA), gelatine and antibody solution. The shape of the fluidic chambers is cut with a programmable cutting machine into the adhesive, which is protected by plastic liners on both sides. One liner is removed and the adhesive is attached to a microscope slide. Fig. 2 illustrates the geometry. We typically use five chambers on one 25 × 76 mm2microscope slide.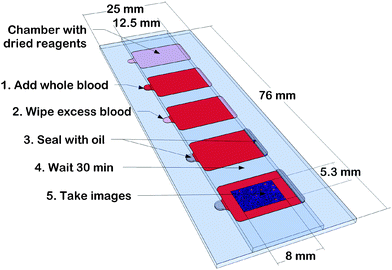 | ||
| Fig. 2 Illustration of the fluidic chip and its usage. Height (26.5 μm) and shape of the test chambers are defined by laminating adhesive between a standard size and half a microscope slide, leaving openings to let blood in and air out. One of the inner surfaces is coated with dried gelatine containing fluorochrome-conjugated antibodies for cell staining. The slide fills by capillary flow, is sealed with mineral oil and after 10–30 min incubation, fluorescence images are taken for cell counting. The inset represents a 0.4 mm × 0.27 mm section of the overlay of two 8 mm × 5.3 mm images after background subtraction. | ||
A solution of 0.15% w/v gelatine is prepared by dissolving gelatine (type A, bloom approx. 300) in micro-filtered deionised (milliQ) water at 40 °C and letting it cool to room temperature. We then add CD3-APC and either CD4-PerCP or CD8-PerCP antibody solution (all from Exbio, Vestec, Czech Republic) to obtain antibody concentrations of 1 ng μl−1 (CD3-APC, IgG2a, Clone MEM-57), ∼0.3 ng μl−1 (CD4-PerCP, IgG1, Clone MEM-241) and 0.5 ng μl−1 (CD8-PerCP, IgG2a, Clone MEM-31), respectively.
This solution is sonicated for ∼5 s to dissolve aggregated antibodies and spun at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g for 10 min to remove remaining aggregates of antibodies. The chamber areas (∼130 mm2) that were previously cut into the tape are filled with 20 μl of the reagent solution and dried at room temperature. This creates gelatine layers with a nominal thickness of 100–150 nm (assuming that the gelatine is completely dry and compact) containing roughly 109antibodies mm−2. After this first drying step, the slides are dipped for 10 s into milliQ water (room temperature) to wash away the antibodies, which are quickly released from the reagent layer. We assume that antibodies in large pores within the collagen network of the gelatine and antibodies close to the surface of the layer are removed in this step. To prevent further release of antibodies, the layer is dried immediately after this washing step (within a few seconds) with dry air. The tape liner is removed and the cover, a microscope slide cut in half (12.5 mm × 76 mm), is attached to define the chamber. Using interferometry we have measured the thickness of several chambers both with and without the reagent layer. The measured chamber height is 26.5 μm ± 1 μm. The resolution of about 0.2 μm is not sufficient to measure significant differences between the heights of coated and uncoated chambers.
000 g for 10 min to remove remaining aggregates of antibodies. The chamber areas (∼130 mm2) that were previously cut into the tape are filled with 20 μl of the reagent solution and dried at room temperature. This creates gelatine layers with a nominal thickness of 100–150 nm (assuming that the gelatine is completely dry and compact) containing roughly 109antibodies mm−2. After this first drying step, the slides are dipped for 10 s into milliQ water (room temperature) to wash away the antibodies, which are quickly released from the reagent layer. We assume that antibodies in large pores within the collagen network of the gelatine and antibodies close to the surface of the layer are removed in this step. To prevent further release of antibodies, the layer is dried immediately after this washing step (within a few seconds) with dry air. The tape liner is removed and the cover, a microscope slide cut in half (12.5 mm × 76 mm), is attached to define the chamber. Using interferometry we have measured the thickness of several chambers both with and without the reagent layer. The measured chamber height is 26.5 μm ± 1 μm. The resolution of about 0.2 μm is not sufficient to measure significant differences between the heights of coated and uncoated chambers.
Imaging setup and image analysis
A schematic representation of the image cytometer is illustrated in Fig. 3. It consists of an LED excitation module, a microscope slide holder and a monochrome CCD camera with a combination of two macro-lenses. The excitation module is taken from an RGB projector (Samsung SP-P410ME).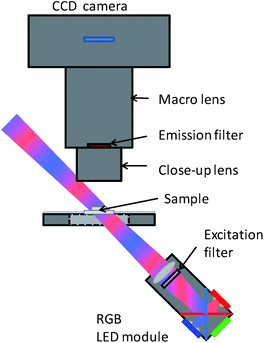 | ||
| Fig. 3 Schematic of the imaging setup. | ||
The contained 5.4 mm2 red, green and blue LEDs were replaced by 4.0 mm2 LEDs from the same manufacturer (Luminus Devices, Phlatlight CBT-40) (red: Popt ≈ 1.6 W, green: Popt ≈ 1.2 W, blue: Popt ≈ 2.2 W) to reduce power consumption and heat generation. The module contains a collimation lens for each LED and combines the optical paths of the three colours with dichroic beam splitter plates. Through a 650 nm short pass filter (Semrock, Rochester, NY, USA) and a lens (f ≈ 30 mm, part of the LED projector), the LED areas are projected with about 3.5× magnification towards the imaging area. Since the optical path of the excitation light is tilted ∼45° with respect to the image plane, this image is distorted and therefore has a slightly non-uniform intensity distribution across the image area. However, the intensity variation is very much smaller than the variation of antigen density recognized by the antibodies on the surface of the cells and is therefore negligible. The fluorescence is collected with a close-up lens (f = 40 mm, LM-scope), filtered with a 685/40 nm band pass filter (Semrock), and imaged onto the chip of the CCD camera (SBIG, ST-1603ME) with a 60 mm macro-lens (Nikon, AF Micro-Nikkor 60 mm f/2.8D). The numerical aperture is mainly limited by the iris diaphragm of the lens, which is set to f/11. The focus ring of the lens is set such that the image on the 13.8 × 9.2 mm2 (1530 × 1020 pixels of 9 × 9 μm2) CCD chip has a magnification of ∼1.7×. The temperature of the CCD chip is kept at −5 °C by a thermoelectric cooler.
As described above we seal the facets of the fluidic chip with oil after filling it with whole blood. After the incubation time, we take one image with red and one with blue excitation light.
The image analysis software is written as a platform independent ImageJ macro. After background subtraction, regions of interest (ROIs) are identified in the sum of the two images. To be identified as a cell, an ROI has to meet several criteria, including the maximum value, the noise (variance) in the neighbourhood and the size and roundness of the area. We use the integrated intensity (after background subtraction) within an ROI for red (blue) excitation as the APC (PerCP) fluorescence intensity. Compensation could easily be applied. However, since we do not apply gates on single parameters, compensation is not useful in a two-parameter measurement.
To characterize the imaging system we have used a variety of fluorescent microbeads. Fluidic chambers built as described above (without the reagent layer) are filled with beads suspended in phosphate buffered saline (PBS). In Fig. 4, the fluorescence intensities of flow cytometry calibration microbeads (Accudrop, APC, PerCP, unlabelled polystyrene beads, all from Becton Dickinson, San Jose, CA, USA) and multi-fluorophore “Rainbow” calibration particles (Spherotech RCP-30-5, Lake Forest, IL, USA) for red and blue excitation are compared. Panel (a) and (b) show scatter plots of the detected intensities of beads suspended in PBS and whole blood (90% whole blood with 10% bead suspension), respectively. The integration time was chosen according to the brightness of the beads between 0.2 s (Accudrop beads, red excitation) and 20 s (unlabelled beads, both colors).
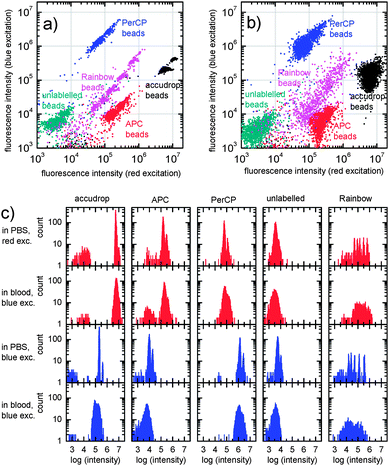 | ||
| Fig. 4 Scatter plots (a and b) and histograms (c) of the fluorescence intensities of microbeads (Accudrop beads, APC, PerCP, unlabelled beads and Rainbow calibration particles) suspended in PBS (panel a; 1st and 3rd row of panel c) and whole blood (panel b; 2nd and 4th row of panel c). | ||
The intensity histograms are displayed in panel (c). For beads suspended in PBS, the coefficients of variation (CV) range between 6% for Accudrop beads excited with the blue LED and 20% for the unlabelled beads with red excitation. Most CVs are around 12%.
In the case of microbeads in whole blood, the surrounding red blood cells lead to a decrease of the fluorescence intensity for blue excitation light of about 30–50%, whereas the intensities for red excitation light do not change considerably. The CVs typically increase to about 25% (red) and 30% (blue), ranging from 20% (APC, red excitation) to 44% (Accudrop, blue excitation).
By comparing the measured intensities with the calibrated intensities of the Rainbow calibration particles and with the intensities of stained lymphocytes, we estimate that the image cytometer can resolve roughly 1000 PerCP or APC molecules in a clear (PBS) solution. In whole blood, about 5000 molecules are needed. This is sufficient to distinguish between blood cells expressing CD3, CD4, or CD8.
Results and discussion
CD4 and CD8 T-lymphocyte counting
T-Lymphocytes express about 200000 CD3 antigens, CD4+ T-lymphocytes about 100000 CD4 antigens and CD8+ T-lymphocyte expression ranges from 30000 to ∼500000 CD8 antigens. Given the results described above, the system should resolve cells labelled with 1:1 conjugates (or higher fluorochrome/antibody ratios) of CD4-PerCP, CD8-PerCP and CD3-APC. Fig. 5 shows scatter plots of the fluorescence intensities of the automatically identified ROIs for red and blue excitation of a test chip with CD4-PerCP/CD3-APC (left) and CD8-PerCP/CD3-APC (right) after filling with whole blood from a finger prick and 30 min incubation at 37 °C. In the scatter plots, the CD4+ CD3+, CD4−CD3+, CD8+ CD3+ and CD8−CD3+ T-lymphocytes can be clearly identified.
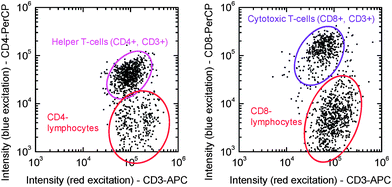 | ||
| Fig. 5 Fluorescence intensities of the automatically identified objects in images from a CD4/CD3 slide (left) and a CD8/CD3 slide (right) after 30 min incubation with whole blood from a finger prick. | ||
To benchmark our approach against flow cytometry, we sampled blood from three donors† in EDTA collection tubes. For flow cytometry, 20 μl of the multi-test reagent CD3-FITC, CD4-APC, CD8-PE, and CD45-PerCP (BD Biosciences) were added to 50 μl of whole blood in a tube containing a known number of fluorescent microbeads (BD Trucount).
After 15 min incubation, the erythrocytes were lysed with 450 μl of a lysing buffer (BD FACSLyse) for another 15 min.
Since, patient blood with low CD4 counts is not easily available in the Netherlands (fortunately, HIV patients receive good treatment with ART), we simulate low lymphocyte counts by creating leukocyte-depleted blood samples. For this, we spin part of the blood samples down (1000 g, 10 min), remove the buffy coat containing most of the leukocytes, and mix the remaining plasma and erythrocytes with different amounts of the initial blood sample to prepare blood with reduced leukocyte concentrations but similar erythrocyte concentrations as the original blood samples. With blood from three donors we prepared samples with nominal concentrations of 100%, 1/3, 1/6, and 0% of the initial concentration. Fig. 6 shows the comparison of the number of CD4 and CD8 T-lymphocytes detected in these samples obtained using the described slide method with flow cytometry (FACSAria II, Becton Dickinson). Cell counting was carried out with previously defined fixed gates in both methods.
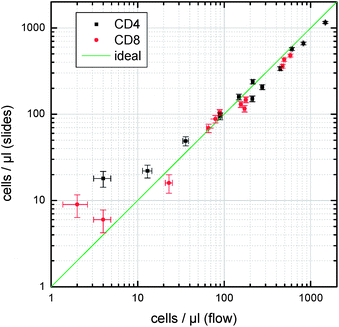 | ||
| Fig. 6 Comparison between lymphocyte concentrations of whole blood samples and leukocyte depleted samples from three donors, measured with the slide-based lymphocyte counting compared with flow cytometry. Error bars indicate the errors expected from Poisson statistics (slides: 1.1 μl of whole blood; flow cytometry: 5 μl). | ||
As can be seen in Fig. 6, the results compare very well over a large range of cell concentrations. With the chosen parameters in the image analysis, we typically miss about 10–15% of the cells (false negatives) and count about 5–10 false positives per slide. We expect that improved algorithms (e.g.cluster analysis instead of statically defined gates) will help to further improve on sensitivity and specificity.
Thermal stability
For a test of the long-term thermal stability of the reagents in the gelatine layer, slides with CD3-APC/CD4-PerCP and CD3-APC/CD8-PerCP reagent coatings were stored together with dry silica gel granules in the dark in airtight containers at room temperature (RT ≈ 20 °C) and at 40 °C. After storage times of up to 15 weeks, fluorescence intensities of CD3+ (red excitation), CD4+ (blue excitation) and CD8+ T-cells (blue excitation) in finger prick blood of healthy donors were obtained from images taken after 45 min incubation at 40 °C. The results are summarized in Fig. 7. The fluorescence intensity reduces at a slow rate (half life: 100–200 days) for the slides stored at room temperature. Most cell counts are within a reasonable range. For the slides stored at 40 °C, we observe a faster degrading of the fluorescence intensity (50–100 days half life) and on several slides, even after a short storage time, the cells were hardly detectable.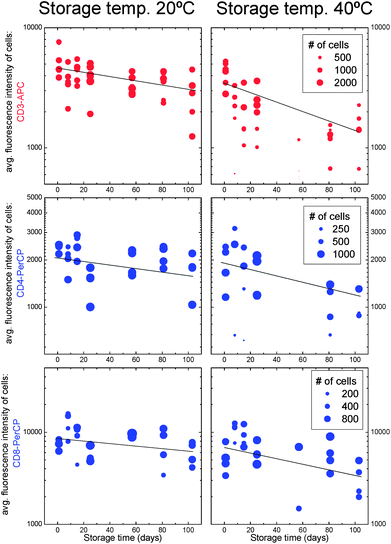 | ||
| Fig. 7 Average fluorescence intensities of cells stained with CD3-APC, CD4-PerCP and CD8-PerCP antibodies after storage of the reagent-coated slides at room temperature (left) and 40 °C (right). The number of counted cells (using manual gating in this case) is represented by the size of the dots. | ||
The cell counts were not compared with other methods. Reference ranges for healthy donors are 1000–2000 (CD3+) T-cells, 500–1500 CD4+ T-cells, and 300–800 CD8+ T-cells.5
This result indicates that a shelf life (half life of the reagent) of at least 50–100 days at 40 °C is possible. However, the large variation between the slides has to be reduced by better quality control during the slide fabrication process.
Alternative protocol: CD4/CD14 for reduced incubation time
The relatively long incubation time of 30 min was used to keep the reagent concentration and therefore the background intensity low. Due to the presence of red blood cells, the background is very non-uniform. For higher reagent concentrations, gaps between red blood cells become indistinguishable from stained cells, and the number of false positives increases. Since these gaps contain both APC and PerCP labelled antibodies, we cannot further increase the reagent concentrations using the combination of CD3 and CD4 to exclude monocytes. We can instead replace CD3 by CD14 in order to discriminate between CD4+ T-cells and CD4(+) monocytes. While CD3 is present on all T-cells, CD14 is expressed by monocytes. Fig. 8 illustrates the advantage of the CD4/CD14 approach over the CD3/CD4 approach for high reagent concentrations. While false positives from background variations interfere with CD4+ T-cells in the case of CD3/CD4, there is no overlap between the cells of interest and the false positives for the combination CD4/CD14.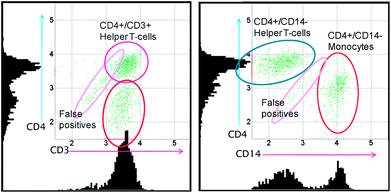 | ||
| Fig. 8 Comparison between the scatter plots and histograms of typical results of CD3-APC/CD4-PerCP tests (left) and CD4-PerCP/CD14-APC tests (right) optimized for fast staining. Results were obtained from images taken after 10 min incubation time. | ||
Similarly, for CD8-counting, CD3-APC can be replaced by a mix of CD16-APC and CD56-APC for the discrimination of natural killer cells from CD8+ T-lymphocytes.
Blood from eight healthy donors was used to compare this method with flow cytometry. From each blood sample, two additional leukocyte depleted samples were prepared as described above with nominal leukocyte concentrations between 10% and 50% of the initial concentrations. The CD4 counts of these 24 samples obtained with the CD4/CD14 slides (CD14-APC: IgG1, Clone MϕP9, BD Biosciences, San Jose, CA, USA) after 10 min incubation at room temperature are compared in Fig. 9 with results using the flow cytometry method described above. Although the deviations between the methods are slightly larger compared with the CD3/CD4 slides incubated for 30 min at 37 °C (Fig. 6), the results indicate that 10 min incubation at room temperature may be sufficient with a further optimized reagent coating.
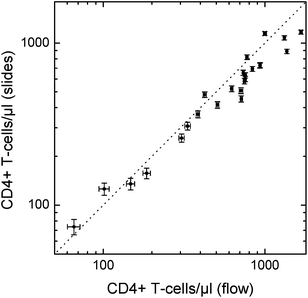 | ||
| Fig. 9 Comparison between cell concentrations obtained using the CD4/CD14 protocol after 10 min incubation on the slides with flow cytometry results. | ||
Identification of antibodies with higher affinities may allow for even shorter incubation times without further increasing the concentrations.
Conclusions
Compared with previous image cytometry approaches, three main innovations make the test simpler and potentially cheaper.First, the optical system in the image cytometer is simplified by the choice of APC and PerCP as fluorochromes. Since one common wavelength range for fluorescence emission can be used, chromatic aberrations do not affect the results and no filter changer is needed. Second, the combination of large-area (>40 mm2) imaging with a thin counting chamber (∼25 μm) ensures sufficient sample volume without the need for sample dilution or RBC lysis. Third, and most importantly, the sample preparation is simplified through the dried reagent layer on the microfluidic chip, which does not require any active mixing, since diffusion across the 25 μm chamber height is sufficient. The slides can be stored without cooling for several months. Since no precision pipetting is required, minimal training will be sufficient to carry out the test in almost any setting, including remote areas in Sub-Saharan Africa, by non-professionals. Timed release of reagents stored in hydrogel layers can also be used to eliminate active mixing in other applications, especially on disposable microfluidic chips. The concept of delayed release is particularly useful where the concept of functionalised surfaces has its drawbacks. Capturing cells on functionalised surfaces typically requires an additional washing step and often sophisticated microstructures are needed to ensure the contact between cells and surface.
The results we present here indicate that on-chip leukocyte staining with antibodies stored in gelatine works well and that it can be used for simple CD4-counting in low-resource settings. With regard to training requirements, this may be one of the simplest approaches for CD4 counting that has been described. A reliable comparison of our approach with state-of-the-art methods will have to be carried out with patient blood including low CD4 counts, for example in Sub-Saharan Africa. Before such tests can be carried out, the slide fabrication procedure needs to be automated and the image cytometer has to be integrated into a portable, battery-operated instrument.
Acknowledgements
This work was funded by the Dutch technology foundation STW under grant number 06146.Notes and references
- J. L. Fahey, J. Giorgi, O. Martínez-Maza, R. Detels, R. Mitsuyasu and J. Taylor, Annales de l'Institut Pasteur / Immunologie, 1987, 138, 245–252 CrossRef CAS.
- M. Badri, S. Lawn and R. Wood, BMC Infect. Dis., 2008, 8, 89 CrossRef.
- D. M. Moore, A. Awor, R. Downing, J. Kaplan, J. S. G. Montaner, J. Hancock, W. Were and J. Mermin, JAIDS, J. Acquired Immune Defic. Syndr., 2008, 49, 477–484 CrossRef.
- O. Keiser, P. MacPhail, A. Boulle, R. Wood, M. Schechter, F. Dabis, E. Sprinz, M. Egger and ART-LINC Collaboration of the International Databases to Evaluate AIDS (IeDEA), Trop. Med. Int. Health, 2009, 14, 1220–1225 CrossRef.
- M. Bofill, G. Janossy, C. A. Lee, D. Macdonald-Burns, A. N. Phillips, C. Sabin, A. Timms, M. A. Johnson and P. B. A. Kernoff, Clin. Exp. Immunol., 1992, 88, 243–252 CrossRef CAS.
- F. Mandy, G. Janossy, M. Bergeron, R. Pilon and S. Faucher, Cytometry, Part B, 2008, 74, S27–S39 CrossRef.
- L. Thairu, D. Katzenstein and D. Israelski, AIDS Care: Psychological and Socio-medical Aspects of AIDS/HIV, 2011 Search PubMed.
- D. S. Boyle, K. R. Hawkins, M. S. Steele, M. Singhal and X. Cheng, Emerging technologies for point-of-case CD4 T-lymphocyte counting, Trends Biotechnol., 2011 DOI:10.1016/j.tibtech.2011.06.015.
- J. P. Golden, J. S. Kim, J. S. Erickson, L. R. Hilliard, P. B. Howell, G. P. Anderson, M. Nasir and F. S. Ligler, Lab Chip, 2009, 9, 1942–1950 RSC.
- P. Kiesel, M. Bassler, M. Beck and N. Johnson, Appl. Phys. Lett., 2009, 94, 041107 CrossRef.
- X. Mao, S.-C. S. Lin, C. Dong and T. J. Huang, Lab Chip, 2009, 9, 1583–1589 RSC.
- P. Kiesel, J. Martini, M. Beck, M. Huck, M. Bern and N. Johnson, Laser Focus World, 2010, 46, 47–50 Search PubMed.
- P. Kiesel, M. Beck and N. Johnson, Cytometry, Part A, 2011, 79, 317–324 CrossRef.
- C. D. Chin, V. Linder and S. K. Sia, Lab Chip, 2007, 7, 41–57 RSC.
- W. R. Rodriguez, N. Christodoulides, P. N. Floriano, S. Graham, S. Mohanty, M. Dixon, M. Hsiang, T. Peter, S. Zavahir, I. Thior, D. Romanovicz, B. Bernard, A. P. Goodey, B. D. Walker and J. T. McDevitt, PLoS Med., 2005, 2, e182 CrossRef.
- S. Mtapuri-Zinyowera, M. Chideme, D. Mangwanya, O. Mugurungi, S. Gudukeya, K. Hatzold, A. Mangwiro, G. Bhattacharya, J. Lehe and T. Peter, JAIDS, J. Acquired Immune Defic. Syndr., 2010, 55(1), 1–7 CrossRef.
- X. Cheng, D. Irimia, M. Dixon, K. Sekine, U. Demirci, L. Zamir, R. G. Tompkins, W. Rodriguez and M. Toner, Lab Chip, 2007, 7, 170–178 RSC.
- X. Cheng, Y.-s. Liu, D. Irimia, U. Demirci, L. Yang, L. Zamir, W. R. Rodriguez, M. Toner and R. Bashir, Lab Chip, 2007, 7, 746–755 RSC.
- N. N. Watkins, S. Sridhar, X. Cheng, G. D. Chen, M. Toner, W. Rodriguez and R. Bashir, Lab Chip, 2011, 11, 1437–1447 RSC.
- Cell Assay Kit and Method, US Pat., application 20090148869, 2009.
- A. Ymeti, X. Li, B. Lunter, C. Breukers, A. G. J. Tibbe, L. W. M. M. Terstappen and J. Greve, Cytometry, Part A, 2007, 71, 132–142 CrossRef.
- X. Li, A. Ymeti, B. Lunter, C. Breukers, A. G. J. Tibbe, L. W. M. M. Terstappen and J. Greve, Cytometry, Part B, 2007, 72, 397–407 CrossRef.
- X. Li, C. Breukers, A. Ymeti, B. Lunter, L. W. M. M. Terstappen and J. Greve, Cytometry, Part B, 2009, 76, 118–126 CrossRef.
- X. Li, C. Breukers, A. Ymeti, K. Pattanapanyasat, K. Sukapirom, L. W. M. M. Terstappen and J. Greve, Cytometry, Part B, 2010, 78, 31–36 Search PubMed.
- V. Hessel, H. Löwe and F. Schönfeld, Chem. Eng. Sci., 2005, 60, 2479–2501 CrossRef CAS.
- I. Barbulovic-Nad, M. Lucente, Y. Sun, M. Zhang, A. R. Wheeler and M. Bussmann, Crit. Rev. Biotechnol., 2006, 26, 237–259 CrossRef CAS.
- J. Moorthy and D. J. Beebe, Lab Chip, 2002, 2, 76–80 RSC.
- C. D. Chin, T. Laksanasopin, Y. K. Cheung, D. Steinmiller, V. Linder, H. Parsa, J. Wang, H. Moore, R. Rouse, G. Umviligihozo, E. Karita, L. Mwambarangwe, S. L. Braunstein, J. van de Wijgert, R. Sahabo, J. E. Justman, W. El-Sadr and S. K. Sia, Nat. Med., 2011, 17, 1015–1019 CrossRef CAS.
- G. Thomas, E. M. El-Giar, L. E. Locascio and M. J. Tarlov, Hydrogel-Immobilized Antibodies for Microfluidic Immunoassays, in Microfluidic Techniques, ed. S. D. Minteer, Humana Press, 2006, vol. 321, pp. 83–95 Search PubMed.
- J. T. Santini, M. J. Cima and R. Langer, Nature, 1999, 397, 335–338 CrossRef CAS.
- B. Roger, et al. , J. Micromech. Microeng., 2008, 18, 075036 CrossRef.
- L. Yobas, L. Feng Cheow, K.-C. Tang, S.-E. Yong, E. Kye-Zheng Ong, L. Wong, W. Cheng-Yong Teo, H. Ji, S. Rafeah and C. Yu, Biomed. Microdevices, 2009, 11, 1279–1288 CrossRef CAS.
- A. Dudia, A. Koçer, V. Subramaniam and J. S. Kanger, Nano Lett., 2008, 8, 1105–1110 CrossRef CAS.
- M. Zimmermann, P. Hunziker and E. Delamarche, Microfluid. Nanofluid., 2008, 5, 395–402 CrossRef.
- M. Hitzbleck, L. Gervais and E. Delamarche, Lab Chip, 2011, 11, 2680–2685 RSC.
- P. Gupta, K. Vermani and S. Garg, Drug Discovery Today, 2002, 7, 569–579 CrossRef CAS.
- R. Langer and J. Folkman, Nature, 1976, 263, 797–800 CrossRef CAS.
- K. Thoma and K. Bechtold, Eur. J. Pharm. Biopharm., 1999, 47, 39–50 CrossRef CAS.
- A. C. Peacock and C. W. Dingman, Biochemistry, 1968, 7, 668–674 CrossRef CAS.
Footnote |
| † Blood donors were healthy individuals at the University of Twente that provided written informed consent. All experiments were performed in compliance with the relevant laws and institutional guidelines. |
| This journal is © The Royal Society of Chemistry 2012 |