A low cost point-of-care viscous sample preparation device for molecular diagnosis in the developing world; an example of microfluidic origami†
A. V.
Govindarajan
,
S.
Ramachandran
,
G. D.
Vigil
,
P.
Yager
and
K. F.
Böhringer
*
University of Washington, Electrical Engineering, Campus Box 352500, Seattle, WA, USA 98195. E-mail: karlb@u.washington.edu; Fax: +1-206-543-3842; Tel: +1-206-221-5177
First published on 8th November 2011
Abstract
The lab-on-a-chip concept has led to several point-of-care (POC) diagnostic microfluidic platforms. However, few of these can process raw samples for molecular diagnosis and fewer yet are suited for use in a resource-limited setting without permanent electrical infrastructure. We present here a very low cost paper microfluidic device for POC extraction of bacterial DNA from raw viscous samples—a challenge for conventional microfluidic platforms. This is an example of “microfluidic origami” in that the system is activated by folding; demonstrated here is room temperature cell lysis and DNA extraction from pig mucin (simulating sputum) spiked with E. coli without the use of external power. The microfluidic origami device features dry reagent storage and rehydration of the lysis buffer. We demonstrate DNA extraction from samples with a bacterial load as low as 33 CFU ml−1. Extraction times, starting from the raw sample, have been optimized to about 1.5 h without the use of external power, or to within 1 h using an oven or a heater block. The fabrication of this paper microfluidic device can be translated into high volume production in the developing world without the need for a semiconductor clean room or a microfabrication facility. The sample preparation can be performed with the addition of just the sample, water, ethanol and elute buffer to the device, thus reducing chemical hazards during transport and handling.
Introduction
The development of nucleic acid amplification tests (NAATs) has opened the doors for highly specific molecular diagnosis and personalized medicine. These tests are currently available in advanced healthcare facilities and are limited in their distribution by their cost and by the lack of point-of-care (POC) sample preparation systems. Simple sample preparation will bring down the cost of molecular diagnosis and make it more accessible. Our goal is two-fold: i) to develop an extremely inexpensive, self-contained, disposable, portable nucleic acid extraction device and ii) to use it to demonstrate extraction of bacterial DNA from a viscous sample matrix, which presents a challenge for conventional microfluidic devices.We describe here the development of a simple paper microfluidic platform for sample preparation. Sample preparation is the first step to nucleic acid-based diagnosis; our long-term goal for the technology shown in this paper is to simplify this complex process such that the cost of nucleic acid extraction will eventually be less than US$2 per patient.
Since the lab on paper review article1 published in 2008, low cost paper diagnostics,2–13 including those based on enzyme-linked immunosorbent assay (ELISA)2, sequential reagent delivery,3 multiplexed bioassays,8 and the integration of paper microfluidics with commercial electrochemical readers14 have been emerging for diagnostic applications in the developing world. Such devices must be inexpensive, rugged, lightweight, and independent of supporting infrastructure.10 Despite advances in conventional and paper microfluidic technology, the lack of development in sample preparation systems has led to there being few lab-on-chip15–19/lab-on-paper5,6 systems that can extract nucleic acids (NA) from raw samples.
We describe our sample preparation platform as an example of “microfluidic origami” because it is fabricated from a stack of flat polymer sheets and paper, and is activated by folding, Demonstrated here is room-temperature cell lysis and DNA extraction from pig mucin (simulating sputum) spiked with E. coli without the use of external power.
A preliminary version of this platform without dry reagent storage was presented at the IEEE International Conference on Microelectromechanical Systems.20 This paper introduces an integrated POC platform that can be operated with the addition of just water, ethanol and elute buffer to the device. This low cost origami-based sample preparation method in combination with an existing or to be developed portable NAAT based system should yield a raw sample-to-result molecular diagnosis in less than 2 h. As a stand-alone device, our approach enables storage of point-of-care extracted DNA for transport at room temperature to more centralized diagnostic facilities. This will be especially useful in the absence of reliable cold shipment or a portable NA amplification system—a typical situation in rural areas of the developing world. This work is the first step towards providing a low cost, low power alternative to a fully automated and portable sample preparation module of an integrated system like the Cepheid GeneXpert.21 The GeneXpert uses self-contained, single use, biosafe, microfluidic cartridges that feature ultrasonic lysis of the filter-captured organisms to release DNA.22
The core idea of this microfluidic origami sample preparation device (Fig. 1) is sequencing complex chemical and physical processing steps through sequential folding of pieces of a flat substrate. This folding sequence puts different paper fluidic circuits in contact with new solvent and reagent sample pads, substituting capillary flow for pumps, and folding and unfolding for valve activation. This microfluidic origami device separates the process into steps on several layers (L0–L4) that fold above or below the central layer L0, which consists of the core elements—the DNA binding filter and a dry lysis buffer storage pad. The device in its present form is disposable and is designed as a single use, inexpensive device that can process one sample at a time.
 | ||
| Fig. 1 Microfluidic origami device for point-of-care nucleic acid extraction. A. Front side, B. Backside, 1. DNA filter, 2. Waste absorption pad with Mylar backing, 3. Sample loading cup, 4. Lysis/wash buffer storage and rehydration pad. 5. Buffer transport channel. 6. Contact stack. L1…L4: Layers that fold above and below layer L0. Figure reprinted from Govindarajan et al.2 | ||
Materials
Sample matrix and bacteria
Commercial mucin (M1778 mucin from porcine stomach, type III, bound sialic acid 0.5–1.5%, partially purified powder from Sigma-Aldrich) was used to simulate sputum. A sample of 20% (w/v) pig mucin in nuclease-free water resembled human sputum in viscosity, stickiness and color. However, the mucin can contain traces of E. coli. To ensure that the extracted and amplified DNA was from the bacterial cell lysis rather than the sample matrix, a transformed E. coli strain, XL1 blue transformed with plasmid DNA H127KSApuc18, was used to spike the mucin in our experiments.Device fabrication and assembly
A single device fabrication (Fig. 2) took less than 30 min. The fabrication consists of stacking layers of paper, Mylar (Fraylock, Inc., San Carlos, CA, USA) and repositionable adhesive, followed by laser cutting. The approach of making both through cuts and partial cuts (by varying the laser power) in combination with peeling of a repositionable adhesive layer presents a low cost alternative to the FLASH method11 for defining paper fluidic channels on a non-wicking substrate. The stacking of layers of the origami from top to bottom was done by hand using a rolling pin to press the sheets together. The origami stack layers from top to bottom comprised of cellulose paper (Millipore, Billerica, MA, USA), 0.5 mil Mylar with double-side adhesive, repositionable adhesive layer (RoyalBrites window decals, Norwalk CT), 4 mil Mylar with single side adhesive and card paper to enable bidirectional folding.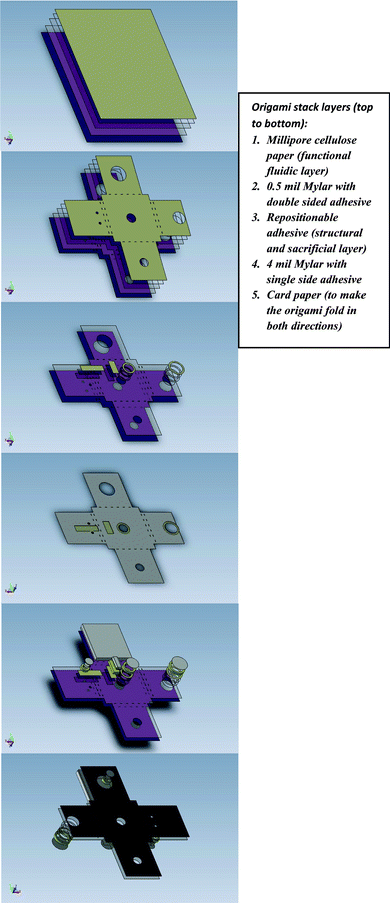 | ||
| Fig. 2 Microfluidic origami NA extraction device fabrication. A. Stack layering, B. Stack through cuts, C. Stack partial cuts, D. Peeling between repositionable window decal and 4 mil Mylar layer to define paper fluidic channels on a non-wicking substrate. The repositionable adhesive film served both as a structural and sacrificial layer. After laser cutting with varying power, the repositionable film was peeled away from the substrate, leaving behind the entire stack in the lower power cut areas, E. Front side assembly of pre-cut parts (with laser cut 3M Scotch mounting tape, serving as spacer), F. Assembling pre-cut parts on backside. Laser cut 3M Scotch mounting tape and Millipore cellulose paper was used to form the sample-loading cup to accommodate larger sample volumes. | ||
The entire stack was placed in a laser cutting machine (M-360 laser engraving and cutting system, Universal Laser Systems Inc, Scottsdale AZ, with a 35 watt CO2 laser). The device was laser cut using a single AutoCAD mask with higher power for the through cuts (power = 40, speed = 10, dpi = 1000, 2 passes) and lower power (power = 28, speed = 10, dpi = 1000, 1 pass) for the partial cuts. The units for the laser cutter settings are (power = % of max. power, i.e. 35 watt, speed = % of max. speed, i.e. 26.5 in s−1, spatial resolution in dots per inch (dpi)).
Partial cuts were made from the top through the repositionable adhesive layer. After laser cutting, the repositionable adhesive layer was peeled from the bottom Mylar and card paper substrate. This left the entire stack (Millipore cellulose paper - card paper) in the regions that were cut with the lower power, and only the substrate (card paper and bottom Mylar layer) in the remaining areas. With this method we were able to fabricate fluidic channels on a non-wicking substrate down to a width of 1 mm.
The remaining parts of the origami device were laser cut and assembled by hand. The adhesive and spacers were formed by laser cutting 3M Scotch mounting tape (3M, St. Paul, MN, USA) (power = 40, speed = 10, dpi = 1000, 3 passes). Millipore cellulose paper was used to form the base of the sample-loading cup and the contact stack, bound by the 3M Scotch mounting tape for the sides of the cup and the contact stack. The Millipore cellulose paper was laser cut with parameters power = 10, speed = 10, dpi = 1000, 3 passes. The DNA capture filter was formed from Fusion 5 membrane (Whatman Inc., Florham Park, NJ) (laser cut with power = 7, speed = 7, dpi = 1000, 1 pass). The waste absorption pad (highly absorbent sterile sponge from Dukal Corporation, Hauppauge NY, catalog# 6125) was hand cut in 2′′ × 2′′ squares. The waste absorption pad was stapled onto the origami device on layer L1, with a contact stack placed in the center so that the waste absorption pad maintains capillary contact with the DNA binding filter. The dry reagent storage pad was made from hand cutting an absorbent 100% pure cotton non-woven fabric (Webril Handi-Pads from Fiberweb, Simpsonville, SC).
Dry lysis buffer storage
For dry reagent storage, wet lysis buffer (composition: 4.5M guanidinium thiocyanate, 50 mM MES (2-(4-morpholino)-ethane sulfonic acid) pH 5.5, 20 mM EDTA, 1% N-lauryl sarcosine (sodium salt), 5% Triton X-100 and 0.25M NaOH, developed in house) was pipetted onto the buffer storage pad. The pad was weighed after addition of the wet lysis buffer, dried in a vacuum oven at 50 °C for about 4 h and stored in a 45 °C incubator until needed (to simulate high temperature environments in the resource limited settings). The last step of device assembly includes placing the dry lysis buffer storage pad on the origami device using 3M Scotch mounting tape.In our laboratory, the fabrication and the operation of the microfluidic origami device were performed in different dedicated spaces with the idea of translating the device production and operation to the developing world while keeping the associated costs to a minimum. We discuss these conditions and make recommendations for high-volume production in a separate section on avoiding contamination.
Experimental
Microfluidic origami device operation
The operation of the microfluidic origami device for NA extraction is described in Fig. 3. (Please also refer to the supplemental video†). The procedure is modified from our earlier presentation20 to include rehydration of dry stored lysis buffer. First a fresh DNA binding filter was placed on the microfluidic origami device and the waste containment pad with the contact stack (on L1) was folded below the filter. Next, the dry lysis buffer storage pad (on L0) was rehydrated with nuclease-free water (please note that the supplemental video shows rehydration off the origami, however we have performed several instances of rehydration on the origami device itself). The difference in the weights of the buffer storage pad after addition of the wet buffer and after drying was used to calculate the volume of water to be added for rehydration. Approximately 490 μl of water was added per 900 μl of wet lysis buffer for rehydration. The dry lysis buffer was rehydrated for approximately 5 min. The raw sample (E. coli-spiked mucin) was then loaded into the sample loading cup (on layer L2, that folds directly above the filter). Next, the buffer transport channel (made of Millipore cellulose paper on L3) was folded above L2. The transport channel made contact with the rehydrated lysis buffer storage pad and the raw sample that was loaded into the cup. A contact stack was included in the design of the transport channel to ensure that the lysis buffer reached the bottom of the sample loading cup, as the viscous sample “decongested” (dissolved with no visible mucin chunks in the sample loading cup) during lysis. | ||
| Fig. 3 Microfluidic origami operation: raw sample - DNA extraction 1. Place fresh DNA binding filter on origami device (A). 2. Fold waste absorption pad below the filter and place lysis buffer storage pad/rehydrate buffer (B). 3. Fold sample loading cup above DNA binding filter and load sample into the cup (C). 4. Fold buffer transport channel (D). 5. Place weight on closed microfluidic origami device to ensure contact and capillarity during lysis operation (∼ 30 min) (E). 6. Unfold origami and cut and discard used/unwanted layers (F). 7. Wash DNA capture filter directly with 100% ethanol (G). 8. Cut off waste absorption pad (H). 9. Dry the DNA capture filter, e.g. on benchtop (I). 10. Elute DNA from filter in low salt solution (J). Note: The microfluidic origami is a low cost disposable device designed to process one sample per use. Gloves were changed after each use. Please refer to the supplemental video.† | ||
The lysis reagents flowed through the temporary fluidic pathways without having to move the sample laterally through the device, thus maximizing the DNA extraction efficiency. At this point, complete (but as-yet-unoptimized) lysis of the sample takes roughly 30 min. A weight was placed on the device in its closed form during lysis to ensure contacts and capillary wicking through layers L0–L3. L4 is reserved for integration of future functionality and was not in use here. After lysis, the weight was removed and the layers were unfolded to expose the DNA binding filter on L0, with the waste containment pad still folded beneath it. Since this is a paper device, the used layers L2 and L3 and the unused L4 layer were at this point cut off and discarded in biohazard waste. This was done to facilitate ease of operation and safety from exposure to harsh chemical residues that resulted from chemical lysis. The DNA binding filter was then washed with 100% ethanol to remove the protein and other non-nucleic acid debris that could be bound to the filter. After the ethanol wash the filter was left to dry (for example, at room temperature).
DNA elution and PCR analysis
After drying, the filter with the bound dry DNA was transferred with a pair of sterile tweezers into a sterile syringe. A 150 μl volume of low salt elute buffer (0.01 M Tris buffer, pH 8) was introduced into the syringe and siphoned up and down, past the DNA filter, using a sterile petri dish at the bottom. The filter was allowed to soak in the elute buffer in the syringe for 5 min before the extracted DNA was collected and stored in a nuclease-free Eppendorf tube, ready for amplification by polymerase chain reaction (PCR). The elutants from the microfluidic origami contained both plasmid and genomic DNA (extracted by a common procedure) that were amplifiable by a conventional benchtop PCR machine (Perkin Elmer Cetus DNA Thermal Cycler).We used malB gene amplification product of 86 bp as a marker for E. coli genomic DNA. For genomic DNA amplification the forward and reverse primers were EC3F: 5′-GCCGATGCC AAATCGTCAG-3′ and EC3R: 5′-GGCGAATACCCAGCGACAT-3′. The PCR used for amplifying genomic DNA incorporated an initial heat step at 95 °C for 3 min followed by 38 cycles of 95 °C for 10 s, 60 °C for 10 s and 72 °C for 15 s. Forward and reverse primers used for the amplification of plasmid DNA were S29: 5′- TCC CAG TCA CGA CGT - 3′ and S33: 5′- AGC CGA TAA CAA TTT CAC ACA GCA - 3′. The PCR thermal process for plasmid DNA consisted of an initial heating step at 96 °C for 2 min followed by 40 cycles of 96 °C for 1 min, 52 °C for 2 min and 70 °C for 3 min (to detect low bacterial loads). For samples spiked with undiluted bacterial culture, 27 cycles were sufficient for detection of the extracted plasmid DNA. The positive controls for PCR were Qiagen (Qiagen Inc. Valencia, CA, USA) extracts from the E. coli culture (genomic extraction control: QIAmp DNA Mini kit, plasmid extraction control: QIAprep Spin Miniprep kit). The positive controls were processed without mucin. The negative control was nuclease-free water. For the plasmid extraction the “mucin only” lane served as an additional negative control. PCR products were separated using agarose gel electrophoresis and were visible in UV light with ethidium bromide staining.
Bacterial load variation
The bacterial DNA extraction experiments to validate the functioning of our device were from E. coli cultures. The E. coli was grown overnight at 37 °C in a shaker bath in Luria-Bertani (LB) broth containing carbenicillin. E. coli cells containing the plasmid DNA H127KSApuc18 are resistant to carbenicillin. In order to test the lower limit of detection sensitivity with the microfluidic origami extraction platform, a fresh culture was grown overnight and was serially diluted in 10 fold steps. A sample of 20 μl of each dilution was plated on LB-agar plates (with carbenicillin) and grown overnight in a 37 °C oven to estimate the colonies grown on the plates. This was translated in terms of colony forming units per ml (CFU ml−1) of the grown culture. 50 μl of each E. coli dilution was spiked in separate 250 μl aliquots of porcine mucin. Each of these spiked raw mucin samples was processed separately on a microfluidic origami device.Avoiding contamination
The fabrication of the microfluidic origami based sample preparation device can be translated in high volumes to the developing world without a microfabrication laboratory (referred to commonly as “cleanroom”) – a typical environment for the fabrication of most lab-on-a-chip systems. This cuts down significantly the associated costs in maintaining the specialized cleanroom equipment and environment (e.g. the particle count). For this work, the origami device was fabricated in a dedicated room that housed a laser cutter, an attached computer and a work bench for assembly. The work bench was cleaned with ethanol and isopropyl alcohol (IPA) prior to hand-assembling laser cut portions of the origami device. The DNA binding filter in particular was stored in a nuclease free Eppendorf tube, immediately after laser cutting the factory sealed and shipped Fusion 5 membrane (Whatman Inc., Florham Park, NJ). It was placed on the origami device just before operation. For more commercial type production and disease diagnosis, assembling the laser cut portions of the device in a laminar flow bench fitted with UV light and high-efficiency particulate air (HEPA) filters would significantly reduce bacterial contamination. During device operation, nuclease free Eppendorf tubes/containers and sterile disposable sample applicators/syringes/tweezers should be used for any intermediate sample or DNA binding filter handling. In our laboratory protocol our dedicated sample preparation space was wiped with ethanol and IPA before processing a batch of samples. A fresh disposable diaper pad was used below to contain any waste or possible spills. Tweezers/sample applicators were sterilized between samples by washing the tips with ethanol. Rinsing in DNase away, RNase away solutions (Invitrogen, Carlsbad, CA) prevented NA degradation. Personal protective equipment included gloves (to be changed for every sample), labcoats, goggles, hairnets and facemasks.Results and discussion
The sample size used in this work was 300 μl of pig mucin spiked with E. coli. The origami-based POC sample preparation device is designed to accommodate larger viscous sample sizes that are readily available from the source and useful to detect lower or sparser bacterial loads. Sample volumes can be downscaled, the limiting factor of fluid flow being the minimum channel width achievable by our fabrication method, which is currently 1 mm. Reagent volumes are yet to be optimized, but we have observed that 2 × 900 μl volumes of lysis buffer are sufficient to dissolve 300 μl of viscous pig mucin. To facilitate ease of manual operation and to accommodate realistically useful viscous sample sizes, our channel widths and lengths and filter and cup diameters are of the order of 1–3.8 cm. We use the term microfluidic because we rely on small-scale physical effects like capillarity and have channel thicknesses and layer heights in the hundreds of μm range.The lysis buffer dissolves mucin at room temperature without external power. It provides complete lysis of the E. coli bacterial cells and does not inhibit PCR. The presence of a specific 500 bp plasmid DNA band, seen after DNA extraction and amplification only in E. coli-spiked mucin samples and not in unspiked mucin samples (Fig. 4, Table 1) confirmed E. coli cell lysis and DNA extraction from the spiked mucin matrix using the microfluidic origami platform. Comparison between lanes 2–4 (extraction with rehydration of dry lysis buffer on the origami) and lane 5 (extraction with direct wet lysis on the origami) in Fig. 4 showed that there was no significant loss in detection due to the buffer rehydration process.
| Lane | 500 bp band expected | 500 bp band seen |
|---|---|---|
| 1. Qiagen kit extraction of plasmid DNA from E. coli | Yes (+ve control) | Yes |
| 2., 3. Mucin + E. coli with dry lysis rehydration (load ∼ 106 CFU ml−1) | Yes | Yes |
| 4. Mucin + E. coli with dry lysis rehydration (load ∼ 104 CFU ml−1) | Yes | Yes |
| 5. Mucin + E. coli with wet lysis | Yes | Yes |
| 6. Mucin only | No | No |
| 7. Water only | No (-ve control) | No |
| 8. 100 bp Invitrogen ladder |
 | ||
| Fig. 4 Extraction of plasmid DNA from raw sample at room temperature, after rehydration of dry stored lysis buffer on microfluidic origami device. Gel lane description given in Table 1. | ||
Fig. 5 (Table 2) shows the extraction and amplification of genomic (naturally occurring) DNA (malB gene of E. coli, 86 bp). For gel images in Fig. 4–Fig. 6 (Table 1–Table 3) note the concurrence in the DNA band “expected” and “seen” columns.
| Lane | 86 bp band expected | 86 bp band seen |
|---|---|---|
| 1. Qiagen kit extraction of genomic DNA from E. coli | Yes (+ve control) | Yes |
| 2., 3. Mucin + E. coli with dry lysis rehydration (load ∼ 106 CFU/ml) | Yes | Yes |
| 4. Mucin + E. coli with dry lysis rehydration (load ∼ 104 CFU/ml) | Yes | Yes |
| 5. Mucin only (mucin is partially purified and may contain traces of E. coli) | Maybe | No |
| 6. Water only | No (-ve control) | No |
| 7. 50 bp Invitrogen ladder |
| Lane | CFU in sample from plating estimate | CFU ml−1 of mucin | 500 bp band expected | 500 bp band seen |
|---|---|---|---|---|
| 1. Qiagen kit extraction of plasmid DNA from E. coli (undiluted culture) | +ve control | Yes | Yes | |
| 2. Mucin + E. coli | 4200 | 1.4 × 104 | Yes | Yes |
| 3. Mucin + E. coli | 485 | 1.6 × 103 | Yes | Yes |
| 4. Mucin + E. coli | 25 | 83 | Yes | Yes |
| 5. Mucin + E. coli | 12 | 40 | Yes | Yes |
| 6. Mucin + E. coli | 10 | 33 | Yes | Yes |
| 7. Mucin + E. coli | 22 | 73 | Yes | Yes |
| 8. Mucin only | No | No | ||
| 9. Water only | -ve control | No | No | |
| 10. 100 bp Invitrogen ladder | ||||
 | ||
| Fig. 5 Extraction of genomic DNA from raw sample at room temperature, after rehydration of dry stored lysis buffer on microfluidic origami device. Gel lane description given in Table 2. | ||
 | ||
| Fig. 6 Bacterial load variation for DNA extraction on the microfluidic origami device. Gel lane description given in Table 3. | ||
The microfluidic origami approach features a raw sample to DNA extraction time (lysis and wash time included) of about 1.5 h without the use of external power or between 45 min–1 h by drying (post lysis and wash) on a heater block at 50 °C or in an oven at 37 °C. In comparison, the raw-sample to extraction time using the FTA® cards5 is as follows: the raw sputum drying on the FTA® card takes 1 h, drying post washes takes another hour, washing with the proprietary FTA® purification reagent and the TE buffer is about 25 min.
Bacterial load variation
Plasmid instead of genomic DNA was used in this work to determine the limits of extraction and detection (after amplification). This is because the mucin used in this work, derived from the porcine stomach, could possibly contain traces of E. coli and would therefore not serve as a good negative background for the bacterial load variation. We acknowledge that diagnosis in a real world setting would be done with genomic DNA and that a closer comparison to existing molecular diagnostic kits would result from using similar bacterial strains of the disease with similar copy numbers. Our goal in this paper is to introduce a low cost proof of concept platform for POC sample preparation leading to POC molecular diagnosis.Fig. 6 (Table 3) is representative of the bacterial load variations in this work. Plasmid DNA bands (500 bp), (after PCR) were visible for the culture, a 10-fold culture dilution and a 100-fold culture dilution. The corresponding bacterial load in the spiked mucin sample as well as an estimate of the colony forming units (CFU) in the actual spiked viscous mucin sample is given in Table 3. These calculations were made from counting bacterial colonies on agar plates.
The concentration of M. tuberculosis bacteria in clinical sputum samples can vary from over 107 to less than 20 CFU ml−1.23 Though our present model is pig mucin spiked with E. coli, our longer-term goal is low cost, POC sample preparation for tuberculosis. We therefore compare our results with standards that are useful for culture-based tuberculosis diagnosis (101–103 bacilli ml−1 of sputum). However, diagnosis of tuberculosis by culture method normally takes 8–12 weeks with additional time for disease subtyping in a national reference laboratory specialized for culturing tuberculosis bacilli from sputum samples. Added to this, culturing requires pretreatment of sputum. The fully automated Cepheid GeneXpert MTB/RIF protocol showed a 95% probability of detecting M. tuberculosis in samples containing a bacterial load of at least 131 CFU ml−1 and was able to detect as few as 10 CFU ml−1 in 35% of the samples22,24 with a raw sample to result time of less than 2 h.22,24 Our goal is to provide a very low cost, and yet effective means to molecular diagnosis that will make disease sub-typing an affordable reality in high disease burden, impoverished areas. Our sample preparation method showed detection by molecular diagnosis for a bacterial load as low as 33 CFU of E. coli ml−1 of raw mucin. This low cost, origami-based sample preparation method in combination with a portable NAAT-based system should yield a raw sample-to-result point-of-care diagnosis in less than 2 h.
Conclusions and future work
We have presented a low cost microfluidic origami device for viscous sample preparation leading to point-of-care (POC) molecular diagnosis. For this DNA extraction device, we set ourselves the following design constraints:1) Tolerance to a viscous sample matrix (mucus or blood).
2) Access to intermediate device layers at different times during operation (in contrast to so-called programmable paper diagnostics12).
3) Ability to process raw samples in a POC setting (similar to other lab-on-paper approaches to sample preparation5,6).
4) Dry reagent storage for cell lysis and penetrating a viscous sample matrix (similar to Guio's approach with FTA® cards5).
5) Raw-sample to DNA extraction time of about 1.5 h without the use of external power or within 1 h using an oven or heater block after the lysis and wash steps.
6) Addition of minimal and safe liquid reagents to the origami device to reduce chemical hazards during transport and handling (in contrast to the protocol described by Guio for the FTA® cards5 that requires the addition of a proprietary FTA® purification reagent and TE buffer, 10 mM Tris HCl, 0.1 mM EDTA pH 8.0, to the chemically impregnated lysis paper).
7) Low cost device made from paper, Mylar and adhesives.
8) Freedom from the need to interface with permanent electrical equipment (vortexer, centrifuge, controlled heater) or Proteinase K (a PCR inhibitor) normally used in NA extraction by commercial kits (e.g. Qiagen).
Our method offers the possibility of POC, room temperature, NA extraction, followed by room temperature storage of the extraction device for transport to more centralized diagnostic facilities. This will potentially reduce the biohazard concerns associated with the shipment of whole samples. Additionally it will eliminate the cost of the cold chain, and sample fixing in the absence of a portable PCR machine.
The fabrication and assembly of this microfluidic origami device takes less than 30 min and can be adapted for high volume production in the developing world without the expensive overhead of a semiconductor cleanroom or microfabrication laboratory facility—a typical environment for the fabrication of lab-on-a-chip devices. All materials used in the fabrication and in the formulation of the lysis buffer are commercially available. The current prototype produces about 150 μl of concentrated nucleic acid solution isolated from a raw sample, ready to be transferred manually to another instrument.
Our sample preparation method allowed detection by molecular amplification from a bacterial load as low as 33 CFU of E. coli per ml of mucin, which falls in the lower limit for culture-based diagnosis of Tuberculosis. This device holds promise for affordable, specific and sensitive POC detection of diseases like Tuberculosis, but we need to move from the E. coli-spiked porcine mucin model system to human sputum loaded with M. tuberculosis, a much more refractory organism. We plan to approach this goal incrementally with the minimum of additional processes on or off the microfluidic origami device. Our origami based NA extraction device is designed to act as a low cost but yet effective front end to a portable NA amplification device. The design and working of the portable amplification and detection device is outside of the scope of this paper.
This particular microfluidic origami device is also being developed as a generic low cost, POC, nucleic acid extraction device for samples of varying viscosities (sputum-urine) and volumes (μl-ml) without the need of permanent electrical infrastructure. We envision that the next generation of microfluidic origami platforms will enable affordable molecular diagnosis and disease sub-typing in geographically dispersed regions with high disease burdens.
Acknowledgements
The authors would like to thank Prof. Patrick Stayton and Dr Richard To for use of the PCR machine, and for donating the transformed XL1-Blue E. coli strain and the primers for the plasmid amplification. We thank Dr Richard To, Dr Barry Lutz, Dr Elain Fu, Dr Paolo Spicar-Mihalic and Peter Kauffman for insightful discussions. The primers used in this work for genomic DNA amplification were from previous work done by the Yager group with Micronics, Inc. This work was supported in part by a gift from BigTec Labs, Bangalore, India to KFB and internal funding by the University of Washington to PY and KFB.Notes and References
- S. J. Sia and L. J. Kricka, Lab Chip, 2008, 8, 1982–1983 RSC.
- C.-M. Chenga, A. W. Matrinez, J. Gong, C. R. Mace, S. T. Phillips, E. Carrilho, K. A. Mirica and G. M. Whitesides, Angew. Chem. Int. Ed., 2010, 49, 4771–4774 CAS.
- E. Fu, P. Kauffman, B. Lutz and P. Yager, Sens. Actuators, B, 2010, 149, 325–328 CrossRef.
- E. Fu, B. Lutz, P. Kauffman and P. Yager, Lab Chip, 2010, 10, 918–920 RSC.
- H. Guio, H. Okayama, Y. Ashino, H. Saitoh, P. Xiao, M. Miki, N. Yoshihara, S. Nakanowatari and T. Hattori, Int. J. Tuberc. Lung Dis., 2006, 10, 906–910 Search PubMed.
- S. R. Jangam, D. H. Yamada, S. M. McFall and D. M. Kelso, J. Clin. Microbiol., 2009, 47, 2363–2368 CrossRef CAS.
- P. Kauffman, E. Fu, B. Lutz and P. Yager, Lab Chip, 2010, 10, 2614–2617 RSC.
- A. W. Martinez, S. T. Phillips, M. J. Butte and G. M. Whitesides, Angew. Chem., Int. Ed., 2007, 46, 1318–1320 CrossRef CAS.
- A. W. Matrinez, S. T. Phillips, E. Carrilho, S. W. Thomas III, H. Sindi and G. M. Whitesides, Anal. Chem., 2008, 80, 3699–3707 CrossRef CAS.
- A. W. Matrinez, S. T. Phillips and G. M. Whitesides, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 19606–19611 CrossRef CAS.
- A. W. Martinez, S. T. Phillips, B. J. Wiley, M. Gupta and G. M. Whitesides, Lab Chip, 2008, 8, 2146–2150 RSC.
- A. W. Martinez, S. T. Phillips, N. Zhihong, C.-M. Cheng, E. Carrilho, B. J. Wiley and G. M. Whitesides, Lab Chip, 2010, 10, 2499–2504 RSC.
- J. L. Osborn, B. Lutz, E. Fu, P. Kauffman, D. Stevens and P. Yager, Lab Chip, 2010, 10, 2659–2665 RSC.
- N. Zhihong, F. Deiss, X. Liu, O. Akbulut and G. M. Whitesides, Lab Chip, 2010, 10, 3163–3169 RSC.
- C. J. Easley, J. M. Karlinsey, J. M. Beinvenue, L. A. Legendre, M. G. Roper, S. H. Feldman, M. A. Huges, E. L. Hewlett, T. J. Merkel, J. P. Ferrance and J. P. Landers, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 19272–19277 CrossRef CAS.
- S.-I. Han, K.-H. Han, A. B. Frazier, J. P. Ferrance and J. P. Landers, Biomed. Microdevices, 2009, 11, 935–942 CrossRef.
- J. W. Hong, V. Struder, G. Hang, W. F. Anderson and S. R. Quake, Nat. Biotechnol., 2004, 22, 435–439 CrossRef CAS.
- J. Kim, M. Johnson, P. Hill and B. K. Gale, Integr. Biol., 2009, 1, 574–586 RSC.
- J. Min, J.-H. Kim, L. Youngsun, K. Namkoong, H.-C. Im, H.-N. Kim, H.-Y. Kim, N. Huh and Y.-R. Kim, Lab Chip, 2011, 11, 259–265 RSC.
- A. V. Govindarajan, S. Ramachandran, G. D. Vigil, P. Yager and K. F. Böhringer, presented at the 24th IEEE International Conference on Microelectromechanical Systems(IEEE MEMS′11), Cancun, Mexico, Jan 23–27, 2011, 932–935 Search PubMed.
- Cepheid, GeneXpert System, http://www.cepheid.com/media/files/brochures/GeneXpert%20Brochure_0112-04.pdf, accessed April 10 2011.
- C. C. Boehme, P. Nabeta, D. Hillemann, M. P. Nicol, S. Shenai, F. Krapp, J. Allen, R. Tahirli, R. Blakemore, R. Rustomjee, A. Milovic, M. Jones, S. M. O'Brien, D. H. Persing, S. Ruesch-Gerdes, E. Gotuzzo, C. Rodrigues, D. Alland and M. D. Perkins, N. Engl. J. Med., 2010, 63, 1005–1015 CrossRef.
- M. L. Joloba, J. L. Johnson, A. Namale, A. Morrissey, A. E. Assegghai, R. D. Mugerwa, J. J. Elner and K. D. Eisenach, Int. J. Tuberc. Lung Dis., 2000, 4, 528–536 Search PubMed.
- D. Helb, M. Jones, E. Story, C. Boheme, E. Wallace, K. Ho, J. Kop, H. R. Owens, R. Rodgers, P. Banada, H. Safi, R. Blakemore, N. T. Lan, E. C. Jones-Lopez, M. Levi, M. Burday, I. Ayakaka, R. D. Mugerwa, B. McMillan, E. Winn-Deen, L. Christel, P. Dailey, M. D. Perkins, D. H. Persing and D. Alland, J. Clin. Microbiol., 2010, 48, 229–237 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1lc20622b |
| This journal is © The Royal Society of Chemistry 2012 |