Ultrathin MnO2 nanofibers grown on graphitic carbon spheres as high-performance asymmetric supercapacitor electrodes†
Zhibin
Lei
,
Jintao
Zhang
and
X. S.
Zhao
*
Department of Chemical and Biomolecular Engineering, National University of Singapore, 4 Engineering Drive 4, 117576, Singapore. E-mail: chezxs@nus.edu.sg; Fax: +65-67791936; Tel: +65-65164727
First published on 4th October 2011
Abstract
Growing MnO2 nanofibers on graphitic hollow carbon spheres (GHCS) is conducted by refluxing GHCS in a KMnO4 aqueous solution aimed to enhance the electrochemically active surface area of MnO2. The stoichiometric redox reaction between GHCS and MnO4− yields GHCS–MnO2 composites with controllable MnO2 content. It is found that these ultrathin MnO2 nanofibers are vertically grown on the external surface of the GHCS, yielding a composite electrode showing good electron transport, rapid ion penetration, fast and reversible Faradic reaction, and excellent rate performance when used as supercapacitor electrode materials. An asymmetric supercapacitor cell with GHCS–MnO2 as the positive electrode and GHCS as the negative electrode can be reversibly charged/discharged at a cell voltage of 2.0 V in a 1.0 mol L−1 Na2SO4 aqueous electrolyte, delivering an energy density of 22.1 Wh kg−1 and a power density of 7.0 kW kg−1. The asymmetric supercapacitor exhibits an excellent electrochemical cycling stability with 99% initial capacitance and 90% coulombic efficiency remained after 1000 continuous cycles measured using the galvanostatic charge–discharge technique.
1. Introduction
Carbon-based materials are promising electrodes for electrochemical energy storage due to their unique properties, such as high surface area, tailorable pore size, and good electrical conductivity.1–3 Commercially available supercapacitors using activated carbon as electrodes usually deliver an order of magnitude lower energy density than batteries.4,5 Thus, recent research has been focusing on optimizing the physical and chemical properties of carbon-based electrode materials to improve their specific capacitances.6–8 However, in an aqueous electrolyte, a symmetric supercapacitor could deliver an energy density of <10 Wh kg−1 because of the limited cell operating voltage (<1.0 V).8,9The energy density of a symmetric supercapacitor can be significantly enhanced by incorporating functional groups on the electrode10 or extending the operating cell voltage.11,12 One of the effective methods is to use an organic electrolyte13,14 with a wider voltage window. However, the organic electrolyte generally exhibits a poor ion conductivity in comparison with the aqueous electrolyte,15 thus leading to a low power density. Moreover, the use of organic electrolytes also brings in environmental concerns. On the other hand, the energy density can be significantly improved by configuring asymmetric supercapacitors with an aqueous solution as electrolyte. By using different electrodes that can be operated at different potential windows, an asymmetric capacitor can be designed with cell voltages extending up to 2.0 V in an aqueous electrolyte.16–20
MnO2 has received a great attention when applied as supercapacitor electrode because of its high theoretical pseudocapacitance, low-cost and environmental compatibilities.21 But, MnO2 has a low electrical conductivity (10−5 to 10−6 S cm−1),22 resulting in a poor rate capability. Recent studies have shown that the electrical conductivity and the rate capability of MnO2 can be significantly improved by compositing MnO2 with a carbonaceous material, such as carbon nanotubes (CNTs)22–25 carbon nanofoams26 and graphene.19,27,28 It has been found that the rate performance of the composite electrodes is closely related to the electrical conductivity of the carbon substrate,29,30 while its specific capacitance is largely determined by the crystallographic form, surface area, and macroscopic morphology of MnO2.31–33 Since MnO2 contributes pseudocapacitance through surface Faradic redox reactions, it has been greatly desirable to prepare MnO2 nanostructures with an ultrathin morphology, which can provide a high electrochemically active surface area for Faradic reaction.30 However, controlled synthesis of such ultrathin MnO2 nanostructures on a highly conductive carbon substrate remains a great challenge.34
Herein, we report the ultrathin MnO2 nanofibers, which were successfully grown on graphitic hollow carbon spheres (GHCS) through a simple redox reaction between GHCS and aqueous KMnO4. The MnO2 content in the GHCS–MnO2 composite can be controlled by changing the initial concentration of KMnO4. An asymmetric supercapacitor manufactured with GHCS–MnO2 as the positive electrode and GHCS as the negative electrode can be reversibly charged/discharged at a maximum cell voltage of 2.0 V in a 1.0 mol L−1 Na2SO4 aqueous electrolyte, delivering a higher energy density in comparison with a GHCS symmetric supercapacitor.
2. Experimental section
Sample preparation
The hollow carbon spheres consisting of graphitic nanorods were prepared as described previously with slight modifications.35 Typically, silica spheres with solid core and mesoporous shell were used as template and ferrocene was employed as the carbon precursor. After chemical vapor deposition at 550 °C for 60 min, the silica/carbon composite was calcined in N2 atmosphere at 850 °C for 90 min to improve the graphitization of the carbon. Removal of the silica template was conducted by using a 20% HF solution, followed by washing with copious deionized water and ethanol to eventually yield the GHCS sample.Growth of ultrathin MnO2 nanofibers on the surface of the GHCS was achieved by direct redox reaction between the GHCS and a KMnO4 solution at 70 °C. Typically, in a 100 mL flask, 30 mg of GHCS was ultrasonically dispersed in 50 mL water. Then, a given amount of KMnO4 (the amount was varied to be 30, 50 or 83 mg) was then added. After stirring for 5 min, the temperature of the solution was raised to 70 °C and refluxed for 120 min. The precipitates were collected by filtration, rinsed with deionized water and ethanol for several times before drying at 95 °C for 5 h.
Characterization
X-Ray diffraction (XRD) of samples was collected on a Shimadzu X-6000 X-ray diffractometer at a scan rate of 3° min−1. Nitrogen adsorption/desorption isotherms were measured at 77 K on a Nova 1000. Samples were degassed at 120 °C for 12 h prior to the measurement. The specific surface areas of the samples were calculated using the Brunauer–Emmett–Teller (BET) method with the adsorption data at the relative pressure (P/Po) range of 0.05–0.2. The total pore volumes were calculated at P/Po = 0.99. The pore size distribution (PSD) curves were computed using the nonlocal density functional theory (NLDFT) model assuming a slit pore geometry. The morphology of the samples was observed on a JEOL JSF-6700F field-emission scanning electron microscope (FESEM). High-resolution transmission electron microscopy (HRTEM) images were collected on a field-emission JEOL JEM-2100F at an acceleration voltage of 200 kV. X-Ray photoelectron spectroscopy (XPS) spectra were collected on an AXIS HSI 165 spectrometer (Kratos Analytical) using a monochromatized Al Ka X-ray source (1486.71 eV). Raman spectra were measured on an Acton Raman spectrometer using a 531.4 nm laser as the excitation source.Electrochemical measurement
The working electrode was prepared by mixing an active material (90 wt%) with carbon black (5 wt%) and polytetrafluoroethylene (5 wt%) in water. The viscous slurry was painted onto two pieces of nickel form with an area of 1 cm2 and pressed under a pressure of 500 kg cm−2. The mass loading of the active material was about 5.0 mg cm−2. The electrochemical performance of samples was characterized by using cyclic voltammetry (CV), galvanostatic charge–discharge and electrochemical impedance spectroscopy (EIS) techniques on an Autolab PGSTAT302N electrochemical workstation. Both three-electrode and two-electrode configurations were used to evaluate the capacitive performances of the electrode materials. In the three-electrode system, a Pt foil and a Ag/AgCl electrode were applied as the counter and reference electrodes, respectively. For the two-electrode configuration, a polyvinylidene fluoride membrane filter (Millipore) was used as the separator. In both cases, a 1.0 mol L−1 aqueous Na2SO4 was used as the electrolyte. The galvanostatic measurement of the two-electrode cell was performed with current densities ranging from 0.1 to 10 A g−1. The specific capacitance, C (F g−1), of the electrode material was calculated from the galvanostatic discharge according to the following equation:27,32| C = I × Δt/(ΔV × m), |
3. Results and discussion
Characterization of GHCS
Fig. 1 shows the FESEM images of the templated GHCS prepared at 850 °C. It is seen that all spheres were uniform in size with an average diameter of ∼280 nm (Fig. 1a). The external surface of the spheres was rather coarse, in sharp contrast to the sample prepared previously at a lower temperature.35 A closer observation showed that each carbon sphere was made up of numerous curved and randomly arranged carbon nanorods, forming many cracks as indicated by the arrows (Fig. 1b). The presence of the cracks is desirable as they may provide channels for the diffusion of electrolyte ions into the interior of the carbon spheres. Fig. 1c shows the TEM images of the carbon spheres. The hollow structure with uniform shell thickness of ∼40 nm can be clearly seen. The magnified image (inset in Fig. 1c) confirmed the randomly aggregated carbon nanorods in the carbon shell. A high-resolution image (Fig. 1d) clearly revealed that these nanorods were composed of densely packed graphene sheets of about fifteen layers, with an interlayer distance of about 0.34 nm. The XRD patterns of the carbon spheres (Fig. S1†) displayed an intensive peak at 25.9° and a weak peak at 43.1°, which can be indexed to the (002) and (100) diffractions of graphitic carbon, respectively.36 These results reveal the high graphitization of the GHCS. | ||
| Fig. 1 (a and b) FESEM and (c and d) TEM images of GHCS. | ||
The nitrogen adsorption/desorption isotherms of the GHCS (Fig. S2†) showed an H4 hysteresis loop, suggesting a hollow structure with porous shells.37 In spite of the same carbon precursor used, the elevated temperature used in this work (850 °C) favored the graphitization of the carbon spheres, consequently resulting in a lower specific surface area (964 m2 g−1, Table 1) in comparison with that of the sample prepared at 800 °C (2239 m2 g−1).35 The pore size distribution curve derived from the NLDFT displayed three peaks (Fig. S3†). The two sharp peaks at 1.6 and 3.0 nm indicate the presence of both micropores and mesopores in the carbon shell, while the broad peak ranging from 7 to 20 nm was most probably caused by the cracks as shown in Fig. 1b.
| Samples | Content of MnO2a (wt%) | S BET /m2 g−1 | V total /cm3 g−1 | ESRc × 10−2/Ohm g | Specific capacitance/F g−1 | |
|---|---|---|---|---|---|---|
| Composite | MnO2 | |||||
| a Content of MnO2 in the composite evaluated by assuming the stoichiometric reaction between GHCS and MnO4−. b Specific surface area and pore volume determined from N2 adsorption. c ESR was calculated from the slope of the linear correlation between the voltage and discharge current density. | ||||||
| GHCS | 0 | 964 | 2.24 | 3.87 | 95 | — |
| GHCS–MnO2-37 | 37 | 560 | 1.61 | 3.90 | 125 | 184 |
| GHCS–MnO2-50 | 50 | 270 | 1.14 | 3.89 | 185 | 280 |
| GHCS–MnO2-64 | 64 | 113 | 0.63 | 3.99 | 190 | 245 |
Growth of MnO2 nanofibers on GHCS
Growth of MnO2 nanofibers on the GHCS was achieved by refluxing aqueous KMnO4 solution in the presence of GHCS at 70 °C. The carbonaceous GHCS served as both reducing agent and substrate for the growth of MnO2 nanofibers.38,39 The reaction between GHCS and MnO4− follows the reaction equation:| 4MnO4− + 3C + H2O → 4MnO2 + CO32− + 2HCO3− |
The survey XPS spectra shown in Fig. S4† reveal the presence of Mn, C and O elements in the composite, suggesting the formation of manganese oxides on the GHCS substrate. The Mn 2p spectra shown in Fig. 2a exhibited two characteristic peaks at 653.9 and 642.2 eV, corresponding to the Mn 2p2/3 and Mn 2p1/2 spin–orbit peaks of MnO2, repectively.19 The energy separation of Mn 3s remains almost the same value of 4.68 eV (Fig. 2b), confirming that the Mn4+ ions are dominant in all the products.31,40
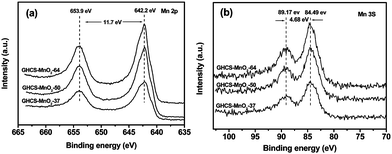 | ||
| Fig. 2 XPS spectra of Mn 2p (a) and Mn 3s (b) of GHCS–MnO2 composites. | ||
The phase of the manganese oxides in the composite was identified by XRD (Fig. S1†). The broad peak at 2θ = 25.9° is clearly due to the GHCS substrate, while the weak peak at 2θ = 12.2° can be indexed to the (001) reflections of birnessite-type MnO2 (JCPDS 42-1317). Based on the Bragg equation, the d(001) value of 0.72 nm can be calculated, which is in good consistence with the birnessite-type MnO2 film grown on graphene28 and CNTs.41 Raman spectra of the composites were also measured to analyze the local structure of MnO2. As shown in Fig. 3, the sharp and strong G bands at 1592 cm−1 relative to the weak D band at 1353 cm−1 confirm a high graphitization of GHCS substrate. In the low wave number region, two weak bands at 573 and 640 cm−1 can also be observed. The former bands are attributed to the Mn–O stretching vibration in the basal plane of MnO6 sheet, while the latter bands are related to the symmetric stretching vibration M–O of MnO6 groups.41,42 In addition, it is known that MnO2 with birnessite phase usually contains hydrated cation in its interlayers.43,44 Thereby, we measured K 2p XPS spectra of GHCS–MnO2 composites (Fig. S5†). The two peaks at 292.6 and 295.3 eV correspond to K 2p3/2 and K 2p1/2, respectively.45 Moreover, with the increase of the MnO2 content in the composite, the K 2p peaks relative to C 1s peaks show a gradual increase in intensity, supporting our conclusion that more birnessite-type MnO2 was loaded on the GHCS at a high concentration of MnO4–.
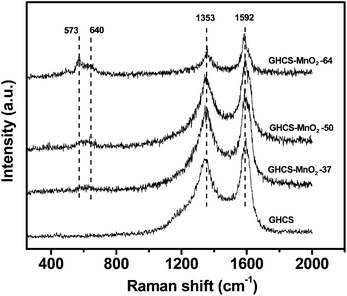 | ||
| Fig. 3 Raman spectra of GHCS and GHCS–MnO2 composites. | ||
Fig. 4a–c show the FESEM images of the GHCS–MnO2 composites. It is seen that all MnO2 shows ultrathin nanofibers that are uniformly distributed on the external surface of GHCS. Moreover, the MnO2 nanofibers show evident increase in number with the increase of MnO4− initial concentrations, confirming a stoichiometric reaction between MnO4− and GHCS. For sample GHCS–MnO2-64 with high MnO2 loading, the GHCS still exhibited a uniform spherical morphology (Fig. 4c) with all spheres covered by ultrathin MnO2 nanofibers.
 | ||
| Fig. 4 FESEM (a–c) and TEM images (d–f) of GHCS–MnO2-37 (a and d), GHCS–MnO2-50 (b, e, and f) and GHCS–MnO2-64 (c). | ||
Fig. 4d–f show the TEM images of the GHCS–MnO2 composite. For sample GHCS–MnO2-37, the MnO2 nanofibers were 20–50 nm in length and sparsely dispersed on the GHCS (Fig. 4d). On increasing the initial concentration of MnO4− (50 mg KMnO4 added in the reaction mixture), both the number and the length of the produced MnO2 nanofibers increased significantly (Fig. 4e). A high-resolution TEM image on the edge of one particle in the sample GHCS–MnO2-50 clearly showed the MnO2 nanofiber and the GHCS sphere (Fig. 4f). The MnO2 phase exhibited a few layers of nanofibers with 5–10 nm in width and less than 100 nm in length. Moreover, a lattice distance of 0.71 nm can be clearly seen, which is in good agreement with d(001) calculated from XRD. Further increasing the amount of KMnO4 to 83 mg in the reaction mixture leads to a significant increase of the number of nanofibers. Moreover, most of these ultrathin MnO2 nanofibers were vertically grown on the external surface of the GHCS (Fig. 5a). The length of MnO2 nanofibers was increased to about 150 nm, while their thickness and width remained not significantly affected by the initial concentration of MnO4− (Fig. 5b). It should be noted that the morphology of MnO2 nanofibers grown on GHCS substrate is entirely different from the previous MnO2 thin film or fibrils, which were coaxially coated on the external surface of the CNT backbone.38,39 In contrast, the vertically grown MnO2 ultrathin nanofibers on the GHCS substrate allow the double sides of MnO2 nanofibers to be accessible by the electrolyte, significantly increasing the electrochemically active surface area of MnO2. Moreover, the HRTEM image of GHCS–MnO2-64 (Fig. S6†) reveals the intimate connections between the MnO2 nanofiber and the conductive GHCS, which are also important for fast electron propagation during the charge–discharge at high current density.22,30 Since MnO4− could diffuse into the interior of GHCS through the cracks as indicated in Fig. 1b, it is anticipated that MnO2 nanofibers should be formed and confined in the interior of the GHCS. Compared to pristine GHCS, the specific surface area of the GHCS–MnO2 composite linearly decreased with the increase of MnO2 nanofibers in the composite (Table 1). However, a broad NLDFT pore size distribution ranging from 7.0 nm to 16 nm (Fig. S3†) suggests that the cracks as indicated in Fig. 1b are still accessible to the exterior. Such open nanostructure allows the electrolyte to diffuse into the interior, enhancing utilization efficiency of MnO2 nanofibers inside the GHCS.
 | ||
| Fig. 5 TEM images of the GHCS–MnO2-64 composite with different magnifications. | ||
Capacitive performances of GHCS–MnO2 composites
The capacitive performance of HCS–MnO2 composites was evaluated by CV and charge–discharge in three-electrode systems. Fig. 6a shows the CV curves of HCS–MnO2 composites at a scan rate of 10 mV s−1 in 1.0 mol L−1 Na2SO4 aqueous electrolyte. The GHCS–MnO2 composites in the potential window of 0–1.0 V exhibit the similar quasi-rectangular CV shapes irrespective of the MnO2 contents in the composite, suggesting that the present MnO2 nanostructure enables the fast access of the electrolyte to the nanofibers even at high MnO2 loading. Fig. 6b shows the galvanostatic charge–discharge curves of the GHCS–MnO2 composite at a current density of 0.1 A g−1. Almost linear shapes with nearly symmetric charge and discharge curves reveal a fast and reversible Faradic reaction between alkali cations (Na+) and the ultrathin MnO2 nanofibers.31,32 The specific capacitance of the composite materials calculated from the galvanostatic discharge process at a current density of 0.1 A g−1 is summarized in Table 1. The low specific capacitance of 95 F g−1 for GHCS is obviously due to its low surface area. Increasing the MnO2 content from 37 to 50 and to 64% leads to a significant increase of the specific capacitance from 125 to 185 and to 190 F g−1, respectively, which is comparable to that of MnO2 nanoflowers grown on CNT arrays (199 F g−1).24 Correspondingly, the capacitance normalized by the pure MnO2 increased from 184 F g−1 to the maximum value of 280 F g−1 and then slightly declined to 245 F g−1 (Table 1). Although the normalized specific capacitance of MnO2 is much lower than its theoretical value of 1370 F g−1 based on the theoretical one-electron redox reaction per manganese atom,31 the specific capacitance of 280 F g−1 reported herein is much higher than that of bulky MnO2 nanorods (180 F g−1),32 nanoflowers (121.5 F g−1),46 nanolamellas (200 F g−1) and other one-dimensional nanostructures (29.3–233.5 F g−1),33 and comparable to that of ultrafine MnO2 nanowire (279 F g−1).47 Such comparison reveals a better utilization efficiency of the present ultrathin MnO2 nanofibers than that of previous MnO2 nanostructures.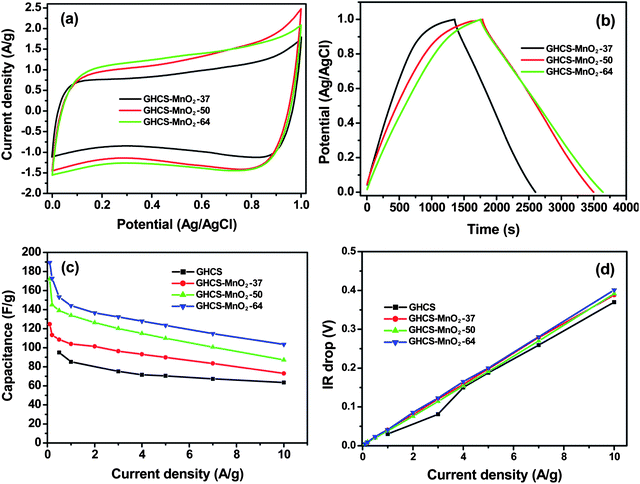 | ||
| Fig. 6 Electrochemical properties of electrode materials measured using a three-electrode system in 1.0 mol L−1 Na2SO4 aqueous electrolyte. (a) CV curves of the GHCS–MnO2 composite recorded at a scan rate of 10 mV s−1, (b) galvanostatic charge–discharge of the GHCS–MnO2 composite at a current density of 0.1 A g−1, (c) variation of specific capacitance against current density and (d) plot of IR drop with discharge current density. | ||
The specific capacitance of the GHCS–MnO2 composite with the change of the current densities is plotted in Fig. 6c. It is seen that the specific capacitance shows a slight decrease with the increase of current density, which reflects a reduced surface area accessed by the electrolyte at a high-current charge–discharge process. Nevertheless, for the GHCS–MnO2-64 composite electrode, its specific capacitance remains as high as 104 F g−1 at a current density of 10 A g−1. The 55% capacitance retention of GHCS–MnO2-64 is in sharp contrast to ∼80% capacitance loss of the MnO2 thin nanofiber electrode coaxially coated on carbon nanofiber arrays (365 vs. 70 F g−1 with current density increased from 0.05 to 15 A g−1).48 The good rate performance of the GHCS–MnO2 composite electrode is clearly attributed to its unique nanostructure. The porous GHCS with high graphitization serves as a highly conductive matrix for fast ion and electron transport, while the ultrathin MnO2 nanofibers on conductive GHCS enable a short diffusion path of electrolyte and provide a more electrochemically active surface area for pseudocapacitance through fast and reversible Faradic reaction. The high performances of the composite electrodes were also reflected from the variation of IR drop with the current density (Fig. 6d). The slopes of linear correlation between the IR drop and discharge current density can be used to evaluate the internal resistance of the electrode materials.49,50 It is interesting to note that the composite electrode exhibits approximately a similar ESR value to pure GHCS (Table 1), suggesting a low internal resistance after MnO2 loading. Such unusual performances suggest that the composite electrode characteristic of the Faradic process behaves more like double-layer capacitance due to the ultrathin MnO2 nanofibers grown on the conductive GHCS.
The EIS of GHCS and GHCS–MnO2 composites and the equivalent circuit diagram used for the fitting of the EIS are displayed in Fig. 7 and in the inset. The equivalent circuit includes bulk solution resistance Rs, the charge transfer resistance Rct, a pseudocapacitive element Cp from the redox process of MnO2 and the constant phase element (CPE) to account for the double layer capacitance. It is seen that all composite electrodes exhibit the nearly vertical line along the imaginary axis in the low-frequency region, revealing an ideally capacitive behavior of the electrode materials due to the fast and reversible Faradic reaction on the ultrathin MnO2 nanofibers. In the high-frequency region (inset in Fig. 7), all the curves intersect the real axis at Rs and Rs + Rct, respectively. After fitting EIS spectra using the equivalent circuit diagram, the bulky solution resistance (Rs) and the charge transfer resistance (Rct) were measured to be 2.1 and 0.9 Ω for GHCS electrode, respectively. The Rs for composite electrodes remained almost the same (ranging from 1.4 to 2.1 Ω) because this parameter is insensitive to the electrode surface, while the Rct was slightly increased to 2.1, 2.6 and 3.5 Ω for GHCS–MnO2-37, GHCS–MnO2-50 and GHCS–MnO2-64 composite electrodes, respectively. However, considering the Rct value (0.9 Ω) of the GHCS substrate and the high mass loading of MnO2, the increase in the magnitude of Rct for the composite electrode is obviously not significant when compared with the MnO2/carbon nanofiber electrode.48 Such low charge transfer resistance is consistent with its good rate performance (Fig. 6c) and low ESR as summarized in Table 1, and thereby suggests a high specific power of the composite electrode. Based on the EIS data, the normalized Cp was calculated to be 60.7 F g−1 for GHCS–MnO2-37, which increased to 66.1 and 87.1 F g−1 for GHCS–MnO2-50 and GHCS–MnO2-64, respectively. Although a direct comparison of Cp with the specific capacitance in Table 1 is not appropriate since they were measured under different conditions, the gradual increase of Cp indeed indicates a more contribution of pseudocapacitance at high MnO2 loading.
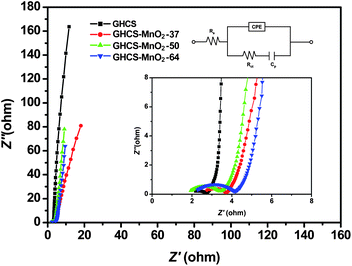 | ||
| Fig. 7 The electrochemical impedance spectra (EIS) of GHCS and GHCS–MnO2 composites with insets showing the high-frequency parts and equivalent circuit diagram used for fitting the EIS. | ||
Asymmetric supercapacitor with GHCS and GHCS–MnO2 as electrodes
To evaluate if the GHCS and GHCS–MnO2 composite can be used to manufacture an asymmetric supercapacitor with a wide potential window, the electrochemical behaviors of GHCS and the GHCS–MnO2-50 composite electrode were measured by a three-electrode configuration with 1.0 mol L−1 Na2SO4 as the aqueous electrode. As shown in Fig. 8, the CV curve of GHCS recorded within a potential window of −1.0 to 0 V (vs. Ag/AgCl) displays a nearly rectangular shape with no obvious gaseous evolution, suggesting a good electrochemical stability of GHCS in negative polarization.51 On the other hand, the GHCS–MnO2-50 electrode is electrochemically stable in the potential window from 0 to 1.0 V (vs. Ag/AgCl). Thereby, it is possible to manufacture an asymmetric supercapacitor with cell voltage extending up to 2.0 V in aqueous electrolyte by using GHCS–MnO2-50 as positive electrode and GHCS as negative electrode.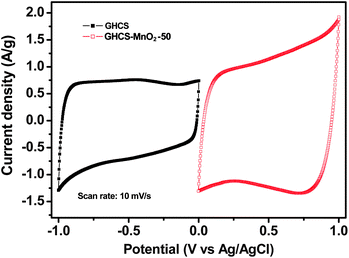 | ||
| Fig. 8 CV curves of GHCS and GHCS–MnO2-50 recorded in a three-electrode cell in 1.0 mol L−1 Na2SO4 aqueous electrolyte at a scan rate of 10 mV s−1. | ||
Fig. 9a shows the CV curves of an asymmetric supercapacitor with different cell voltages varying from 0–1 V to 0–2 V. The mass ratio of negative electrode (GHCS, 10.08 mg) to positive electrode (GHCS–MnO2-50, 5.13 mg) was fixed close to 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, which was based on charge balance theory16,20 and their respective specific capacitances in 1.0 mol L−1 Na2SO4 aqueous electrolyte (95 vs. 185 F g−1 in Table 1). It is seen that the asymmetric supercapacitor exhibits an ideal capacitive behavior with nearly rectangular CV curves even at potential window up to 2.0 V. Moreover, at scan rate as high as 100 mV s−1, the rectangular CV shape can still be well preserved at a maximum cell voltage of 2.0 V (Fig. 9b), suggesting a good rate capability of the asymmetric supercapacitor.
:1, which was based on charge balance theory16,20 and their respective specific capacitances in 1.0 mol L−1 Na2SO4 aqueous electrolyte (95 vs. 185 F g−1 in Table 1). It is seen that the asymmetric supercapacitor exhibits an ideal capacitive behavior with nearly rectangular CV curves even at potential window up to 2.0 V. Moreover, at scan rate as high as 100 mV s−1, the rectangular CV shape can still be well preserved at a maximum cell voltage of 2.0 V (Fig. 9b), suggesting a good rate capability of the asymmetric supercapacitor.
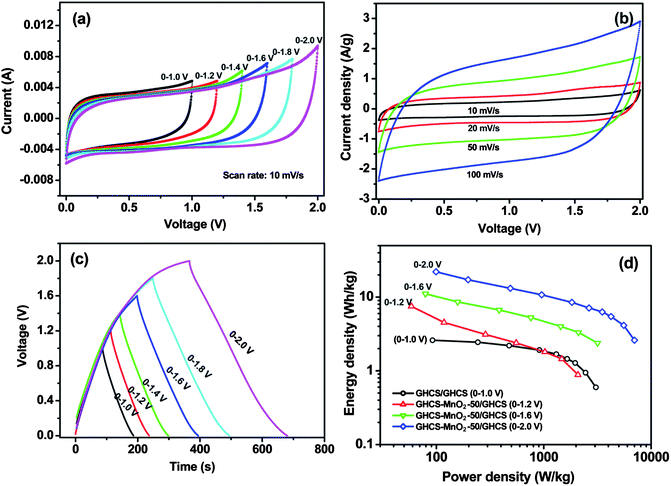 | ||
| Fig. 9 Capacitive performances of an asymmetric supercapacitor with GHCS–MnO2-50 as positive electrode and GHCS as negative electrode. (a) CV curves at different cell voltages, (b) CV curves recorded at different scan rates with a maximum cell voltage of 2.0 V, (c) galvanostatic charge–discharge at a current density of 0.2 A g−1, (d) Ragone plot of the asymmetric supercapacitor showing the relation of energy density and power density. The electrolyte was 1.0 mol L−1 Na2SO4 aqueous solution. | ||
The galvanostatic charge–discharge curve of the asymmetric capacitor at a current density of 0.2 A g−1 is shown in Fig. 9c. It is seen that both charge and discharge curves remain a good symmetry at cell voltage as high as 2.0 V, in sharp contrast to the previous asymmetric supercapacitor with MnO2 and In2O3 nanowires as electrode materials, which exhibited noncapacitive and nonsymmetric behavior as the cell voltage increased above 1.5 V.18 The present excellent capacitive performance at a cell voltage of 2.0 V suggests the long-term stability of the asymmetric supercapacitor. The high cell voltage in aqueous electrolyte affords the supercapacitor high energy density and high power density. Fig. 9d shows the Ragone plot of the asymmetric capacitor charged/discharged at different cell voltages. All data were obtained based on the total mass of the two electrodes' configuration. It is seen that the supercapacitor with symmetric GHCS electrode delivered a much low energy density of 2.6 Wh kg−1 at a power density of 92 W kg−1, due to its low specific capacitance and restricted operating voltage (1.0 V). In contrast, the asymmetric supercapacitor shows significant enhancement in both energy density and power density. It is clearly seen that the energy densities of the asymmetric capacitor are highly dependent on the cell voltages. In particular, at a cell voltage of 2.0 V, the supercapacitor delivered an energy density of 22.1 Wh kg−1 at a power density of 100 W kg−1 and maintained 4.1 Wh kg−1 at a power density of 5.5 kW kg−1. Such an excellent capacitive performance can be ascribed to the use of aqueous electrolyte with high ion conductivities and the synergetic role of the two asymmetric electrodes. The negative electrode of GHCS accumulates charge through double-layer capacitance and provides fast electron transfer, whereas the ultrathin MnO2 nanofibers grown on GHCS provide short diffusion length of electrolyte and more electrochemically active surface area for fast and reversible Faradic reaction.
To investigate the long-term cycling performance of the asymmetric capacitor operating at a maximum cell voltage of 0–2.0 V, we performed the consecutive galvanostatic charge–discharge cycles on an asymmetric supercapacitor with GHCS–MnO2-64 as positive electrode and GHCS as negative electrode. As shown in Fig. 10, the capacitance of the supercapacitor shows a slight decrease in the initial 300 cycles, and then increases and remains quite constant with cycle number. The specific capacitance based on the total mass of the two electrodes after 1000 cycles was measured to be 24.4 F g−1 at a current density of 1.0 A g−1, corresponding to 99% of its initial capacitance (24.5 F g−1). This retention is much better than the previous MnO2-based asymmetric capacitor, such as MnO2–graphene/graphene (79% after 1000 cycles),19 MnO2/activated carbon (93% after 100 cycles),17 MnO2/CNTs (89% after 1000 cycles),22 and comparable to MnO2–graphene/activated carbon nanofibers (97.3% after 1000 cycles).20 Moreover, the coulombic efficiency,52 defined as the ratio of the total amount of discharge to charge calculated from the galvanostatic experiment, remains as high as 90% after 1000 charge–discharge cycles. This excellent electrochemical stability reveals a highly reversible Faradic reaction between the electrolyte and the ultrathin MnO2 nanofibers. Given that the specific capacitance of the negative electrode GHCS is not very high (95 F g−1), the energy density of the asymmetric capacitor is expected to be further boosted by replacing the GHCS with other high-specific-capacitance carbon electrodes. Such efforts are currently underway.
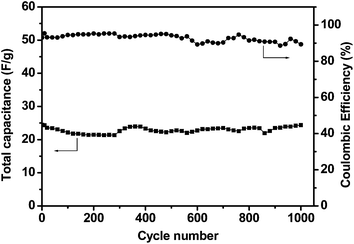 | ||
| Fig. 10 Variation of total capacitance and coulombic efficiency with cycle number for an asymmetric supercapacitor with GHCS–MnO2-64 as positive electrode and GHCS as negative electrode. The cell voltage is 2.0 V and the current density is 1.0 A g−1. | ||
4. Conclusions
Ultrathin MnO2 nanofibers have been uniformly grown on the graphitic hollow carbon spheres by stoichiometric redox reaction between GHCS and MnO4−. The porous GHCS with high graphitization serve as an excellent conductive substrate for rapid electron transfer, while the ultrathin MnO2 nanofibers grown on GHCS provide a large electrochemically active surface area for fast and reversible Faradic reaction. An asymmetric supercapacitor manufactured with GHCS–MnO2 as positive electrode and GHCS as negative electrode can be reversibly charged/discharged at a maximum cell voltage of 2.0 V in 1.0 mol L−1 aqueous Na2SO4 electrolyte, delivering an energy density of 22.1 Wh kg−1 and a power density of 7.0 kW kg−1, much higher than 2.6 Wh kg−1 and 3.0 kW kg−1 of a supercapacitor with symmetric GHCS electrode. Moreover, the asymmetric supercapacitor preserves 99% of its initial capacitance and retains 90% coulombic efficiency even after 1000 cycles of galvanostatic charge–discharge. Such excellent capacitive performance makes the GHCS–MnO2 composite a promising electrode material for the electrochemical energy storage.Acknowledgements
We are grateful for financial support of Singapore Ministry of Education under a Tier 2 Grant (Project no. MOE2008-T2-1-004).Notes and references
- P. J. Hall, M. Mirzaeian, S. I. Fletcher, F. B. Sillars, A. J. R. Rennie, G. O. Shitta-Bey, G. Wilson, A. Cruden and R. Carter, Energy Environ. Sci., 2010, 3, 1238 RSC.
- L. L. Zhang and X. S. Zhao, Chem. Soc. Rev., 2009, 38, 2520 RSC.
- C. Liu, F. Li, L. P. Ma and H. M. Cheng, Adv. Mater., 2010, 22, E28 CrossRef CAS.
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845 CrossRef CAS.
- S. W. Lee, B. M. Gallant, H. R. Byon, P. T. Hammond and Y. Shao-Horn, Energy Environ. Sci., 2011, 4, 1972 RSC.
- Z. B. Lei, N. Christov, L. L. Zhang and X. S. Zhao, J. Mater. Chem., 2011, 21, 2274 RSC.
- Y. Korenblit, M. Rose, E. Kockrick, L. Borchardt, A. Kvit, S. Kaskel and G. Yushin, ACS Nano, 2010, 4, 1337 CrossRef CAS.
- D. W. Wang, F. Li, M. Liu, G. Q. Lu and H. M. Cheng, Angew. Chem., Int. Ed., 2008, 47, 373 CrossRef CAS.
- Z. B. Lei, N. Christov and X. S. Zhao, Energy Environ. Sci., 2011, 4, 1866 RSC.
- S. W. Lee, N. Yabuuchi, B. M. Gallant, S. Chen, B. S. Kim, P. T. Hammond and Y. Shao-Horn, Nat. Nanotechnol., 2010, 5, 531 CrossRef CAS.
- D. Hulicova-Jurcakova, A. M. Puziy, O. I. Poddubnaya, F. Suárez-García, J. M. D. Tascón and G. Q. Lu, J. Am. Chem. Soc., 2009, 131, 5026 CrossRef CAS.
- C. Peng, S. Zhang, X. Zhou and G. Z. Chen, Energy Environ. Sci., 2010, 3, 1499 RSC.
- M. D. Stoller, S. Park, Y. Zhu, J. An and R. S. Ruoff, Nano Lett., 2008, 8, 3498 CrossRef CAS.
- C. Liu, Z. Yu, D. Neff, A. Zhamu and B. Z. Jang, Nano Lett., 2010, 10, 4863 CrossRef CAS.
- P. Simon and A. Burke, Electrochem. Soc. Interface, 2008, 17, 38 Search PubMed.
- T. Brousse, P. L. Taberna, O. Crosnier, R. Dugas, P. Guillemet, Y. Scudeller, Y. Zhou, F. Favier, D. Belanger and P. Simon, J. Power Sources, 2007, 173, 633 CrossRef CAS.
- M. S. Hong, S. H. Lee and S. W. Kim, Electrochem. Solid-State Lett., 2002, 5, A227 CrossRef CAS.
- P.-C. Chen, G. Shen, Y. Shi, H. Chen and C. Zhou, ACS Nano, 2010, 4, 4403 CrossRef CAS.
- Z.-S. Wu, W. Ren, D.-W. Wang, F. Li, B. Liu and H.-M. Cheng, ACS Nano, 2010, 4, 5835 CrossRef CAS.
- Z. Fan, J. Yan, T. Wei, L. Zhi, G. Ning, T. Li and F. Wei, Adv. Funct. Mater., 2011, 21, 2366 CrossRef CAS.
- W. F. Wei, X. W. Cui, W. X. Chen and D. G. Ivey, Chem. Soc. Rev., 2011, 40, 1697 RSC.
- S. W. Lee, J. Kim, S. Chen, P. T. Hammond and Y. Shao-Horn, ACS Nano, 2010, 4, 3889 CrossRef CAS.
- Y. Hou, Y. W. Cheng, T. Hobson and J. Liu, Nano Lett., 2010, 10, 2727 CrossRef.
- H. Zhang, G. P. Cao, Z. Y. Wang, Y. S. Yang, Z. J. Shi and Z. N. Gu, Nano Lett., 2008, 8, 2664 CrossRef CAS.
- J. Yan, Z. J. Fan, T. Wei, J. Cheng, B. Shao, K. Wang, L. P. Song and M. L. Zhang, J. Power Sources, 2009, 194, 1202 CrossRef CAS.
- A. E. Fischer, K. A. Pettigrew, D. R. Rolison, R. M. Stroud and J. W. Long, Nano Lett., 2007, 7, 281 CrossRef CAS.
- S. Chen, J. Zhu, X. Wu, Q. Han and X. Wang, ACS Nano, 2010, 4, 2822 CrossRef CAS.
- J. Yan, Z. Fan, T. Wei, W. Qian, M. Zhang and F. Wei, Carbon, 2010, 48, 3825 CrossRef CAS.
- H. Jiang, L. Yang, C. Li, C. Yan, P. S. Lee and J. Ma, Energy Environ. Sci., 2011, 4, 1813 RSC.
- L. Bao, J. Zang and X. Li, Nano Lett., 2011, 11, 1215 CrossRef CAS.
- M. Toupin, T. Brousse and D. Bélanger, Chem. Mater., 2004, 16, 3184 CrossRef CAS.
- Q. Qu, P. Zhang, B. Wang, Y. Chen, S. Tian, Y. Wu and R. Holze, J. Phys. Chem. C, 2009, 113, 14020 CrossRef CAS.
- S. Chen, J. Zhu, Q. Han, Z. Zheng, Y. Yang and X. Wang, Cryst. Growth Des., 2009, 9, 4356 CrossRef CAS.
- H. Xia, M. O. Lai and L. Lu, J. Mater. Chem., 2010, 20, 6896 RSC.
- Z. B. Lei, Z. W. Chen and X. S. Zhao, J. Phys. Chem. C, 2010, 114, 19867 CrossRef CAS.
- Z. B. Lei, S. Y. Bai, Y. Xiao, L. Q. Dang, L. Z. An, G. N. Zhang and Q. Xu, J. Phys. Chem. C, 2008, 112, 722 CrossRef CAS.
- M. Kruk and M. Jaroniec, Chem. Mater., 2001, 13, 3169 CrossRef CAS.
- S. B. Ma, K. Y. Ahn, E. S. Lee, K. H. Oh and K. B. Kim, Carbon, 2007, 45, 375 CrossRef CAS.
- X. Jin, W. Zhou, S. Zhang and G. Z. Chen, Small, 2007, 3, 1513 CrossRef CAS.
- M. Toupin, T. Brousse and D. Bélanger, Chem. Mater., 2002, 14, 3946 CrossRef CAS.
- S. B. Ma, Y. H. Lee, K. Y. Ahn, C. M. Kim, K. H. Oh and K. B. Kim, J. Electrochem. Soc., 2006, 153, C27 CrossRef CAS.
- C. Julien, M. Massot, R. Baddour-Hadjean, S. Franger, S. Bach and J. P. Pereira-Ramos, Solid State Ionics, 2003, 159, 345 CrossRef CAS.
- S. Ching, D. J. Petrovay, M. L. Jorgensen and S. L. Suib, Inorg. Chem., 1997, 36, 883 CrossRef CAS.
- J. Liu, V. Makwana, J. Cai, S. L. Suib and M. Aindow, J. Phys. Chem. B, 2003, 107, 9185 CrossRef CAS.
- M. L. Xiang, D. B. Li, J. Zou, W. H. Li, Y. H. Sun and X. C. She, J. Nat. Gas Chem., 2010, 19, 151 Search PubMed.
- J. Ni, W. Lu, L. Zhang, B. Yue, X. Shang and Y. Lv, J. Phys. Chem. C, 2009, 113, 54 CrossRef CAS.
- H. Jiang, T. Zhao, J. Ma, C. Y. Yan and C. Z. Li, Chem. Commun., 2011, 47, 1264 RSC.
- J. Liu, J. Essner and J. Li, Chem. Mater., 2010, 22, 5022 CrossRef CAS.
- M. D. Stoller and R. S. Ruoff, Energy Environ. Sci., 2010, 3, 1294 RSC.
- C. W. Huang and H. S. Teng, J. Electrochem. Soc., 2008, 155, A739 CrossRef CAS.
- L. Demarconnay, E. Raymundo-Pinero and F. Béguin, Electrochem. Commun., 2010, 12, 1275 CrossRef CAS.
- C.-T. Hsieh and H. Teng, Carbon, 2002, 40, 667 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: XRD patterns, XPS spectra of GHCS and GHCS–MnO2 composites, N2 sorption isotherms, NLDFT pore size distributions, HRTEM images of GHCS–MnO2-64. See DOI: 10.1039/c1jm13872c |
| This journal is © The Royal Society of Chemistry 2012 |