Surface phase diagram of hematite pseudocubes in hydrous environments†
Haibo
Guo
* and
Amanda S.
Barnard
Virtual Nanoscience Laboratory, CSIRO Materials Science and Engineering, Clayton, Victoria 3168, Australia. E-mail: haibo.guo@csiro.au
First published on 6th October 2011
Abstract
Hematite nanoparticles often display the pseudocubic morphology enclosed exclusively by the (012) surface. The surface chemistry on these facets is important to understand the formation and properties of these nanoparticles in varying chemical environments. The surface is typically terminated by hydroxyl groups in water or humid atmospheres, and the various terminations differ largely in composition, structure, thermodynamic stability, and chemical reactivity. When we compare a large variety of chemical configurations, we find that three specific terminations are thermodynamically stable under aerobic conditions. The termination by singly and triply coordinated hydroxyl groups (S-,T-OH) is stable at low temperatures and in hydrous environments, the stoichiometric clean termination is stable at high temperatures and in dry environments, and the termination by doubly coordinated hydroxyl groups (D-OH) is stable under intermediate conditions. The S-,T-OH termination can convert topologically to the clean surface by dissociative adsorption of water, whereas the conversion from these two terminations to the D-OH termination requires re-organization of several atomic layers at the surface. Therefore, the surface may reversibly change between S-,T-OH and the clean surface depending on the temperature and humidity. Based on these findings we have constructed surface phase diagrams to predict the termination types in different hydrous and humid environments.
1 Introduction
The surface chemistry of iron oxides in hydrous environments (both liquid water and humid atmospheres) is of significant geochemical and technological importance. In a lot of bio-geochemical processes, the widespread iron oxides are reactive and mobile agents that regulate the transport of nutrients and pollutants, and cycle among a bewildering array of polymorphs.1,2 Because of their natural abundance and unique physical and chemical properties, iron oxides have long been used as pigments, catalysts, absorbents, magnetic recording media, and medical image-contrasting agents.2 Reducing the size of individual particles to nanoscale dimensions has significantly improved their performance and expanded their applications by tuning the chemical, electronic and magnetic properties of iron oxide nanostructures. Once formed, surfaces often host key processes or reactions in natural and anthropogenic environmental settings, and many natural and synthetic nanostructures of iron oxides are formed, stored, or disposed in hydrous environments. Hence, knowledge of the interactions between water and the surfaces not only helps us understand the natural geochemical processes, but also guides us in the synthesis of functional nanostructures for specific applications.Hematite (α-Fe2O3) is one of the most stable phases of iron oxides under ambient conditions.2 It has the corundum-type structure with hexagonal close packed oxygen planes between which Fe3+ occupies 2/3 of the available octahedral sites. Hematite is weakly ferromagnetic between 260 K and 950 K, with the spin moments in neighbouring anion interlayers aligned in anti-parallel directions. This crystal and electronic structures lead to highly anisotropic electrical conductivity.3 The surfaces of hematite also display different hydroxylation configurations, which are relevant to the surface chemistry, protonic acidity, and charge accumulation.4 The combination of the anisotropic bulk and surface properties give rise to a surface potential gradient that triggers electrochemical reactions between (001) and (hk0) surfaces.5 Also this anisotropicity provides opportunities for engineers to optimise the performance for specific applications. In this respect, the photocatalytic6 and photoelectrochemical properties7,8 have been reported to benefit from the conductive basal plane. It is, however, convenient to separately study the effects from the bulk and the surfaces, so that one may optimise their respective properties. For example, hematite photo-anodes have detrimental surface states and low charge diffusion efficiency which may be solved separately.9 It is also desirable to separate the effects of different surfaces, whose structure and composition are sensitive to environmental conditions, during the study of the hematite nanostructures. This is a nontrivial task as it requires accurate control over environmental conditions and surface preparations.
The morphologies of hematite nanoparticles vary largely with environmental conditions and synthesis methods.2 The sol–gel synthesis route often leads to the formation of the pseudocubic nanoparticles which are enclosed exclusively by the (012) surface.10 The pseudocubic hematite nanoparticles can also be synthesized using hydrothermal method,11–13 and forced hydrolysis,14 and has been predicted to be the most thermodynamically stable compared with prism and rhombohedron shapes in anhydrous environments.15 The pseudocube is different from the morphologically anisotropic shapes (e.g.nanorods or nanotubes) that are bounded by slow growing surfaces in addition to the (often roughened) fast growing surface. The pseudocubic shape is actually a rhombohedron with the angle between unit cell edges close to 90°. It is frequently found in corundum-type structures where the c/a of the hexagonal unit cell is around 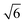 , with which the angle is exactly 90° (see Fig. 1). In hematite the rhombohedrons are enclosed exclusively by one of the surfaces of {101}, {012}, {104}, and {018}. Since the rhombohedrons only expose one type of surface, they respond to changing conditions uniformly, and can therefore be used in gas sensors with high sensitivity, fast recovery, and reproducibility.11
, with which the angle is exactly 90° (see Fig. 1). In hematite the rhombohedrons are enclosed exclusively by one of the surfaces of {101}, {012}, {104}, and {018}. Since the rhombohedrons only expose one type of surface, they respond to changing conditions uniformly, and can therefore be used in gas sensors with high sensitivity, fast recovery, and reproducibility.11
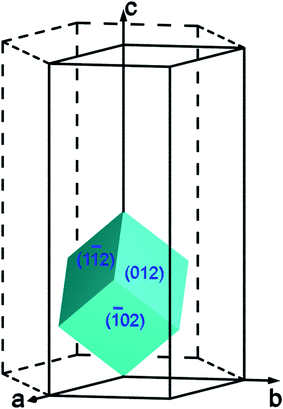 | ||
| Fig. 1 The orientations of the (012) surface in the unit cell of hematite. | ||
Studies of the hematite (012) surface have examined the surface structure and hydroxylation,16–23 as well as surface charge accumulation and proton affinity,24,25 using both experimental and computational techniques. However, the surface stability under diverse environmental conditions (temperatures, partial pressures) is still lacking as these studies mostly focus on specific conditions. Experimental studies encountered difficulties in controlling environmental conditions and characterising the hydrated surfaces in hydrous environment. Computational modelling is advantageous in accurately managing the environmental conditions and control the surface compositions and structures, and have been extensively used in the study to the (001)23,26–29 and (012)16,19,21,23 surfaces of hematite. However, we have shown accurate computational studies of iron oxides are very challenging because of the structural and electronic properties of this complicated system.30 Another complication of the surface is the tremendous possible configurations of hydroxylation, over 1000 in a moderate 2 × 2 supercell.25,31 In this study we will use previously tested30 computational methods and settings, together with the first-principles thermodynamics, to study a reasonable range of surface structures and their stability of the hematite (012) surface.
2 Computational method and settings
The surface energies are calculated using density functional theory (DFT) implemented in VASP (Vienna ab initio simulation package).32,33 We have chosen Generalized Gradient Approximation (GGA) with the exchange–correlation functional of Perdew, Burke and Ernzerhof (PBE).34 The potentials of core electrons and nuclei of Fe, H, and O are reproduced using the projector augmented wave method (PAW)35 with core radii of 2.3, 1.1, 1.52 Bohr, respectively. The PAW potential of Fe is generated with the valence configuration of d7s1, and with nonlinear core correction using a partial-core radius of 2.0 Bohr. Our convergence tests determined that a plane-wave cutoff of 500 eV is sufficient for all the gas molecules and solid structures to converge the calculated energies to 2 meV/atom. We use a Gaussian distribution function for smearing of electronic occupation with a smearing width of 0.1 eV, and extrapolate the total energies to zero electronic temperature.Geometry optimizations are performed using the conjugate gradient algorithm until the residual forces are smaller than 0.01 eV/Å. The convergence criteria for self-consistent electronic iterations are 10−5 eV. The k-points are generated using the Monkhorst–Pack scheme36 and tested for the convergence criteria of 1 meV/atom for both bulk and surface structures. We used a k-point mesh of 11 × 11 × 11 for the rhombohedral unit cell of bulk hematite, and a 5 × 5 × 1 mesh for the surface slabs.
To account for the strong correlation effects we have included the on-site Coulomb interactions with the simplified rotationally invariant formulation by Dudarev et al.,37 where only the effective Coulomb repulsion parameter Ueff = U − J (U is the Coulomb parameter, and J is the Stoner parameter for exchange field) is significant. Our previous tests showed the value of Ueff = 4.5 eV can reproduce lattice parameters of five iron oxides and oxyhydroxides.30
The surfaces are represented by periodic slab models, each slab having two free surfaces bounded by vacuum. The surface vectors are 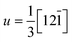 and v = [100] with respect to the hexagonal unit cell of hematite (a = 5.0715 Å and c = 13.8781 Å, see Ref. 30), or u = [001] and v = [1
and v = [100] with respect to the hexagonal unit cell of hematite (a = 5.0715 Å and c = 13.8781 Å, see Ref. 30), or u = [001] and v = [1![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0] with respect to the rhombohedral unit cell. The atoms in the middle of the slabs are frozen at their bulk positions, and the atoms at the two free surfaces are allowed to relax. The use of symmetric slab models is to avoid discontinuities in electric potentials under periodic boundary conditions. As a result we found the surface net dipole moment of the slabs are negligibly small. The thicknesses of the frozen bulk part of the slabs are 6 repeating units of the stacking sequence of the (012) surface, that is the atomic planes of -O-Fe-O-Fe-O- (Fig. 2). The thicknesses of the free surface parts are more than 1 repeating unit according to our convergence tests, as the difference in the calculated surface energies is 1–2 mJ m−2 with more surface layers being allowed to relax. Our convergence tests showed that a vacuum of 10 Å thick is sufficient to reduce the error associated with the interactions with periodic image slabs to less than 2 mJ m−2. The thickness of the slabs is tested with the convergence criteria of 5 mJ m−2. Symmetrization is switched off for the slab models, and dipolar corrections are included.
0] with respect to the rhombohedral unit cell. The atoms in the middle of the slabs are frozen at their bulk positions, and the atoms at the two free surfaces are allowed to relax. The use of symmetric slab models is to avoid discontinuities in electric potentials under periodic boundary conditions. As a result we found the surface net dipole moment of the slabs are negligibly small. The thicknesses of the frozen bulk part of the slabs are 6 repeating units of the stacking sequence of the (012) surface, that is the atomic planes of -O-Fe-O-Fe-O- (Fig. 2). The thicknesses of the free surface parts are more than 1 repeating unit according to our convergence tests, as the difference in the calculated surface energies is 1–2 mJ m−2 with more surface layers being allowed to relax. Our convergence tests showed that a vacuum of 10 Å thick is sufficient to reduce the error associated with the interactions with periodic image slabs to less than 2 mJ m−2. The thickness of the slabs is tested with the convergence criteria of 5 mJ m−2. Symmetrization is switched off for the slab models, and dipolar corrections are included.
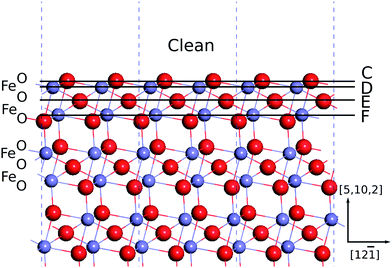 | ||
| Fig. 2 Illustration of surface termination types. A horizontal line indicates a type-X termination is obtained by removing the atoms on and above the line. Oxygen atoms are in red, and iron atoms are in grayish blue. | ||
Based on these calculations we coordinate reference states in the framework of first-principles thermodynamics, which is presented in Ref. 15 together with the thermodynamic parameters, and the chemical potentials of bulk ferromagnetic body-centered cubic Fe and the gases (H2, O2, and water vapour). In first-principles thermodynamics, the difference in chemical potentials at ground state and at standard state is defined to be the connection energy:
| Δμ(Tr,p0) = μ(Tr,p0) − μ(0 K, 0 Pa), | (1) |
| ΔG = Σniμi(T,p) = Σni[Ei(0 K, 0 Pa) + Δμi(T,p)], | (2) |
The connection energy can be computed via an integration of Δμ(T,p0) = ∫T0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) KCp(T′)dT′ − T∫T0KCp(T′)/T′dT′ given that the thermodynamic function of heat capacity is known in the temperature range from 0 K to T, or be calculated using reaction equilibrium in eqn (2) given that free energies of formation are experimentally measured. In the present study we used the latter method with measured formation energies of iron oxides and oxyhydroxides from Ref. 40, and the results of the connection energies of H2, O2, and H2O vapour are presented in Ref. 15. This method was also used to calculate the chemical potential of Fe bulk at standard state, and thus circumvented the difficulty in calculating the energy difference with different U parameters for Fe bulk and for iron oxide and oxyhydroxide bulks.
KCp(T′)dT′ − T∫T0KCp(T′)/T′dT′ given that the thermodynamic function of heat capacity is known in the temperature range from 0 K to T, or be calculated using reaction equilibrium in eqn (2) given that free energies of formation are experimentally measured. In the present study we used the latter method with measured formation energies of iron oxides and oxyhydroxides from Ref. 40, and the results of the connection energies of H2, O2, and H2O vapour are presented in Ref. 15. This method was also used to calculate the chemical potential of Fe bulk at standard state, and thus circumvented the difficulty in calculating the energy difference with different U parameters for Fe bulk and for iron oxide and oxyhydroxide bulks.
It was reported in vacuum the stoichiometric (012) surface can form a (1 × 2) reconstruction which has surface Fe2+ ions.18,20 The (1 × 2) reconstruction is known to have half surface oxygen sites for hydrogenation as the (1 × 1) reconstruction, but its surface structure is not clear.18,20 Therefore, we have limited our discussion to the (1 × 1) reconstruction.
3 Results and discussions
3.1 Configurations of surface hydroxylation
As mentioned above, the stacking sequence of the (012) atomic planes is -O-Fe-O-Fe-O- (Fig. 2). The clean surface is obtained by truncating bulk hematite between the two adjacent oxygen layers. In hydrous environments the (012) surface was found to be capped by an oxygen layer, with the sub-surface Fe layer missing.17 We have included the structures with a sub-surface Fe layer removed from the clean surface, and refer to this termination as A. The clean surface contains triply coordinated oxygen atoms which can be hydrogenated. We have passivated the dangling oxygen bonds, and refer to this termination as B.By removing the atomic layers as shown in Fig. 2 we obtain terminations C–F, among which C and E are terminated by iron atoms, whereas D and F are terminated by oxygen atoms. The termination C has no unsaturated near-surface oxygen atoms. D has doubly coordinated oxygen atoms at the surface layer, and triply coordinated oxygen atoms at the sub-surface layer. E has a layer of triply coordinated oxygen atoms. And F has singly and triply coordinated oxygen atoms. Since each atomic layer contains two atoms, we can construct new termination types by removing half of the surface atoms. These sparse terminations are referred to as G to J, corresponding respectively to the dense terminations C to F. These dangling oxygen bonds are hydrogenated to construct different configurations of hydroxylation. The atomic structures are shown in detail in the Electronic Supplementary Information.
It is noted that the terminations A and F (and its sparse version J) both have two successive oxygen layers, and have both singly and triply coordinated oxygen atoms. The important difference is that A also has doubly coordinated oxygen atoms. Both terminations may be thought of as missing an iron layer, but F can be obtained by simply truncating the bulk hematite.
3.2 Thermodynamic phase diagrams of different terminations
Most of the surface terminations are non-stoichiometric, and accordingly, their surface energies are dependent on the chemical potential and amount of excess components. We calculate the surface energies using| γ = (Eslab − ΣiNiμi)/(2A), | (3) |
The surface energies in Table 1 are calculated using the chemical potentials of H2O and O2 at the thermodynamic standard state. In the scheme of first principles thermodynamics, we have assumed that the energy difference between solids is invariant with pressure and temperature, therefore, the change in surface energies are caused by gas phases, whose chemical potentials change as μ(T,p) = μ(T,p0) + RTln(p/p0). To facilitate the calculation of μH2O we elected to use the gas-phase water (or water vapour) as our starting point to calculate μH2O even though at the thermodynamic standard state water is liquid. This treatment allows us to naturally account for a broad range of conditions, for example, humid gases and ultra-high vacuum, and the chemical potential of liquid water is easily calculated using saturated vapour pressures of water at given temperatures.
| n(H2O) (nm−2) | n(O2) (nm−2) | γ (J m−2) | S (nm−2) | D (nm−2) | T (nm−2) | Description |
|---|---|---|---|---|---|---|
| 0.0 | 0.0 | 1.061 | 0.0 | 0.0/0.0 | 0.0/0.0 | Clean |
| 3.6 | 3.6 | 3.244 | 3.6 | 0.0/7.2 | 0.0/0.0 | AA |
| 7.2 | 1.8 | 1.665 | 3.6 | 3.6/3.6 | 0.0/3.6 | AC |
| 10.8 | 0.0 | 0.730 | 3.6 | 3.6/3.6 | 3.6/3.6 | AE |
| 7.2 | 1.8 | 1.976 | 3.6 | 0.0/3.6 | 3.6/3.6 | AG |
| 3.6 | 0.0 | 0.625 | 0.0 | 0.0/0.0 | 0.0/3.6 | DA |
| 3.6 | 0.0 | 0.770 | 0.0 | 3.6/3.6 | 0.0/3.6 | DB : dense D-OH |
| 7.2 | 0.0 | 0.440 | 3.6 | 3.6/3.6 | 3.6/3.6 | FA : dense S-,T-OH |
| 3.6 | 1.8 | 1.689 | 3.6 | 0.0/0.0 | 0.0/3.6 | FC |
| 3.6 | 1.8 | 1.705 | 3.6 | 0.0/0.0 | 0.0/3.6 | FD |
| 1.8 | −0.9 | 1.379 | 0.0 | 1.8/1.8 | 0.0/3.6 | HB: sparse D-OH |
| 3.6 | −0.9 | 1.466 | 0.0 | 1.8/3.6 | 1.8/1.8 | IA |
| 3.6 | −0.9 | 1.747 | 0.0 | 1.8/3.6 | 1.8/1.8 | IB |
| 5.4 | −1.8 | 1.826 | 0.0 | 3.6/3.6 | 1.8/1.8 | IC |
| 3.6 | −0.9 | 1.482 | 0.0 | 3.6/3.6 | 0.0/1.8 | ID |
| 1.8 | 0.9 | 1.407 | 1.8 | 0.0/0.0 | 0.0/5.4 | JA |
| 1.8 | 0.9 | 1.408 | 1.8 | 0.0/0.0 | 0.0/5.4 | JB |
| 3.6 | 0.0 | 0.841 | 1.8 | 0.0/0.0 | 0.0/3.6 | JC |
| 5.4 | −0.9 | 0.958 | 1.8 | 1.8/1.8 | 1.8/1.8 | JD : sparse S-,T-OH |
| 5.4 | −0.9 | 0.958 | 1.8 | 1.8/1.8 | 1.8/1.8 | JE |
We present the temperature- and pressure-dependent phase diagrams in Fig. 3 and 4 for the temperatures of 298.15 K and 373.15 K, respectively. In the phase diagrams the range of μH2O covers the conditions that can be achieved in UHV chambers, while the range of μO2 are extended to include highly anaerobic conditions which can be achieved by using reductive agents such as H2, N2H4, or CO, which lower μO2 substantially and may reduce hematite into magnetite or iron. The μO2 required to reduce hematite into magnetite is calculated using μ = 6E(hematite) − 4E(magnetite). It is noted that the chemical potential shown in the phase diagrams is always a difference with reference to the specific temperature and under the standard pressure, as Δμ = μ − μ(T,p0). Similarly the μO2 for decomposing bulk hematite into bulk Fe is calculated.
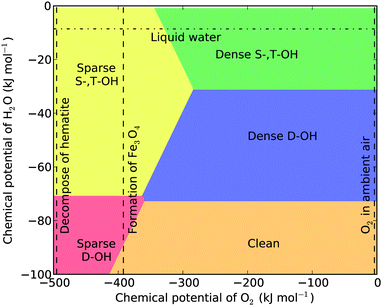 | ||
| Fig. 3 Surface phase diagram of hematite (012) at T = 298.15 K. The three vertical dashed lines mark the chemical potentials of oxygen corresponding to the formation of bulk Fe, the formation of magnetite, and O2 in ambient air. The horizontal dash-dotted line marks the chemical potential of liquid water. | ||
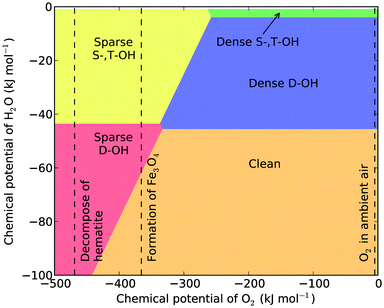
From the phase diagrams we see that except under the highly anaerobic conditions, there are three most probable surface terminations: the dense singly and triply coordinated hydroxylation (dense S-,T-OH), the dense doubly coordinated hydroxylation (dense D-OH), and the clean surface. The phase boundaries among them are independent of μO2, and their presence only depends on μH2O. This is not surprising as their chemical compositions can be written as xFe2O3·yH2O. From the two phase diagrams one sees that the dense S-,T-OH termination occurs mainly in water or humid environments, the clean surface occurs in dry environments, and the dense D-OH termination is stable under intermediate conditions. The surface stability in hydrous environment agrees with the experimental observations that there are mainly singly and triply coordinated hydroxyl groups under hydrous conditions.16,22,31
3.3 Atomic structures of the two hydroxylated surfaces
By comparing the atomic structures of the dense S-,T-OH termination and the dense D-OH termination, we found that the former contains a substantial quantity of hydrogen bonds while the latter has no hydrogen bonds (see Fig. 5). At the dense S-,T-OH termination the hydrogen bonds form continuous rows that are connected at singly coordinated oxygen atoms. This feature is clearly seen in the top view (Fig. 6). The rows of the hydrogen bonds are aligned along the [12] direction. The configuration of hydrogen bonds is very similar as the one proposed in Ref. 16 except that there are no doubly coordinated oxygen atoms in our model. The difference originates from the way that the models are constructed. The model in Ref. 16 was constructed by removing a layer of sub-surface Fe atoms from the clean surface, while our model was constructed by simply truncating the bulk hematite.
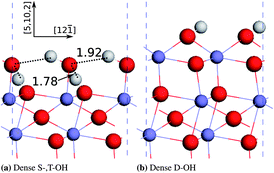 | ||
| Fig. 5 Optimized atomic structures of the dense S-,T-OH termination and the dense D-OH termination. The hydrogen bonds are shown by dashed lines and the bond distances between H and the donor oxygen atoms (unit: Å). Hydrogen atoms are in white colour, oxygen in red, and iron in grayish blue. | ||
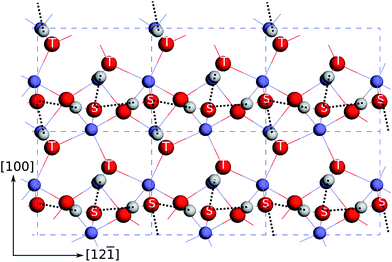 | ||
| Fig. 6 Top view of the atomic structures at the dense S-,T-OH termination. The hydrogen bonds are indicated by the dashed lines. The top surface singly coordinated oxygen atoms are marked with “S”, and the sub-surface triply coordinated oxygen atoms are marked with “T”. See Fig. 5 for the notations of the colours of atoms. | ||
3.4 Temperature dependence of surface hydroxyl groups
Under a wide range of aerobic conditions, the thermodynamic stability of the surface terminations only depends on the chemical potential of water at a given temperature. This is indicated by the horizontal phase boundaries in the surface phase diagrams. Therefore, we can construct a phase diagram to describe these conditions with temperature and μ(H2O) as variables, and μ(O2) as a parameter. We elected to use the chemical potential of oxygen in air under ambient conditions for the parameter, but the phase diagram constructed with this value (Fig. 7) can cover μ(O2) in the range from 0 to −300 kJ mol−1 (where sparse S-,T-OH termination may become thermodynamically stable) at the shown temperatures.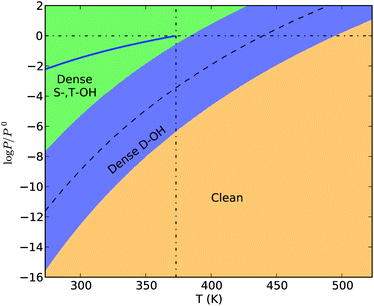 | ||
| Fig. 7 The surface phase diagram of hematite (012) under aerobic conditions. The bold blue line represents the saturated vapour pressure of water in the temperature range from 273.15 K to 373.15 K. The dashed curve is the phase boundary between the S-,T-OH termination and the clean termination. The vertical dash-dotted line marks 373.15 K, the boiling temperature of water under 1 atm. The horizontal dash-dotted line marks the standard pressure (1 atm). | ||
The chemical potential of liquid water (μH2O(l)) at a given temperature is dependent on the solution conditions,41 but for dilute solution conditions we can approximately use the chemical potential of pure water, which equals to the chemical potential of water vapour under the saturated vapour pressure at a given temperature. We have drawn the saturated vapour pressure of water in the temperature range from 273.15 K to 373.15 K in Fig. 7, to show the chemical potential of liquid water (the solid line). One sees the termination with dense singly and triply coordinated hydroxyl groups is thermodynamically stable in liquid water environments at temperatures up to 373.15 K. This type of termination has been proposed to be stable based on geometric arrangements of surface ions and groups, 4 and observed experimentally using X-ray diffraction16 and high electron energy loss spectroscopy.22 Our calculation results confirm the thermodynamic basis for the phase stability.
Pressures larger than 1 atm are required to maintain the liquid state of water above the boiling temperature. Hence under pressures below 1 atm and at temperatures above 373.15 K, water should be supplied to the surface system as a humid gas. If we keep the partial pressure of water vapour (pH2O) constant and increase the temperature, the surface termination will change from dense S-,T-OH termination to the dense D-OH termination and to the clean surface, assuming there is no energy barrier for the surface phase transitions. The transition temperatures depend on the partial pressure of water vapour. For example, when pH2O = 1 atm, the transition temperature for dense S-,T-OH to dense D-OH is approximately 381 K, and for pH2O = 3.2 kPa (saturated vapour pressure of water at room temperature), the transition temperature is approximately 355 K. To initiate the transition to the clean surface, higher temperatures or lower partial pressures of water vapour are required. With pH2O = 1 atm the transition temperature from the dense D-OH to the clean surface is 495 K. Under vacuum conditions the transition temperatures will be substantially reduced. It has been reported that water molecules desorb from hematite (012) surface at 353 K measured using temperature programmed desorption under ultra-high vacuum (UHV).22,31 This desorption temperature is approximately 45 K higher than our calculations for the UHV condition (10−7 Pa or 10−12 atm), suggesting the water desorption requires an activation energy. This phase diagram is therefore useful in predicting the surface terminations under various conditions of temperature and humidity.
In these discussions we have assumed there are no activation energies required for the surface phase transitions, that is, the occurrence of a certain surface phase is solely determined by relative energetic stability under thermodynamic equilibrium conditions. However, surface phase transitions entail rearrangements of surface atoms, adsorption and desorption of gas molecules, and all these processes require activation energies. By analysing the atomic structures of the three terminations, we found the transition from the dense S-,T-OH termination to the clean surface should be facile since it only requires the dissociation of H2O into –OH and –H terminal groups, or the recombination of two terminal groups into H2O molecules. However, the transition between the dense S-,T-OH and the dense D-OH terminations is topologically unfeasible, and requires substantial rearrangement of near surface atoms. The rearrangement might be activated by dissolution-reprecipitation processes in solution environments, but the dense S-,T-OH termination is thermodynamically favourable in solutions. Alternatively, the rearrangement of H and O atoms might be achieved by evaporation-condensation processes in gas environments, which are unlikely to move the surface Fe atoms. To obtain necessary surface diffusion of Fe and O atoms, high temperatures are generally required which favour the formation of the clean surface. Therefore, even though the D-OH is thermodynamically stable under appropriate thermodynamic conditions, it is likely missing from the transition path between the dense S-,T-OH and the clean terminations. In Fig. 7 we also show the calculated phase boundary (the dashed curve) between the dense S-,T-OH and the clean terminations. All the transition temperatures are sensitive to the partial pressure of water vapour.
4 Conclusions
We have constructed surface phase diagrams of hematite (012) under a range of environmental conditions, using the first-principles thermodynamics with inputs from both measured thermodynamic data and our DFT calculations. Unless under highly reductive conditions, the presence of different hydrated and hydroxylated terminations are independent on the chemical potential of oxygen. The dense S-,T-OH termination, dense D-OH termination, and the clean surfaces are thermodynamically stable depending on the chemical potential of water. In water or a humid atmosphere and at low temperatures below 373.15 K, the dense S-,T-OH termination is the most thermodynamically stable phase. At high temperatures the stable termination is likely to be the clean surface. In the intermediate region, both surface terminations are thermodynamically metastable with respect to the dense D-OH termination.The dense S-,T-OH termination has singly and triply coordinated hydroxyl groups, which are more active in charge accumulation than doubly coordinated hydroxyl groups.5 Therefore, pseudocubic hematite nanoparticles that are enclosed by the (012) surface should be more reactive when charge transfer is involved in water environment. Since the change between this dense S-,T-OH termination and the clean termination is topologically facile, the pseudocubic hematite nanoparticles can be synthesized or stored at high temperatures or in dry environment, and re-activated in water or humid atmosphere through dissociative water adsorption. As mentioned in the introduction, since the pseudocubes are enclosed exclusively by the (012) surface, which responds to changes uniformly, they are suitable for gas-sensor applications.
The dense S-,T-OH termination has connected rows of hydrogen bonds, which may be used to transfer protons. The thermal stability of this termination, as shown in the temperature-dependent phase diagram (Fig. 7), may limit their application at high temperatures. However, the energy barrier of water desorption may hold the surface hydroxyl groups at higher temperatures than shown in the equilibrium phase diagram. It was shown water molecules desorb from the (012) surface at 353 K in vacuum,25,31 and the temperature of water desorption in humid atmosphere may suffice for certain applications (e.g. as filler materials in low-temperature proton exchange membranes). The topological inter-conversion between this dense S-,T-OH termination and the clean termination should make them to be activated easily by moist or liquid water.
Acknowledgements
This project has been partially supported by the Australian Research Council under grant number DP0986752. The authors thank Prof. Huifang Xu for fruitful discussions on the pseudocubic morphology, and acknowledge the supercomputer usage and support from the NCI National Facility in Australia, under Merit Allocation Scheme grant p00.References
- M. F. Hochella, Jr., S. K. Lower, P. A. Maurice, R. L. Penn, N. Sahai, D. L. Sparks and B. S. Twining, Science, 2008, 319, 1631–1635 CrossRef.
- R. M. Cornell and U. Schwertmann, The iron oxides, Wiley-VCH, Weinheim, Germany, 2nd edn, 2003 Search PubMed.
- N. Iordanova, M. Dupuis and K. M. Rosso, J. Chem. Phys., 2005, 122, 144305 CrossRef CAS.
- V. Barrón and J. Torrent, J. Colloid Interface Sci., 1996, 177, 407–410 CrossRef.
- S. V. Yanina and K. M. Rosso, Science, 2008, 320, 218–222 CrossRef CAS.
- C. M. Eggleston, A. J. A. Shankle, A. J. Moyer, I. Cesar and M. Grätzel, Aquat. Sci., 2009, 71, 151–159 CrossRef CAS.
- R. H. Goncalves, B. H. R. Lima and E. R. Leite, J. Am. Chem. Soc., 2011, 133, 6012–6019 CrossRef CAS.
- M. Cornuz, M. Grätzel and K. Sivula, Chem. Vap. Deposition, 2010, 16, 291–295 CrossRef CAS.
- F. Le Formal, N. Tétreault, M. Cornuz, T. Moehl, M. Grätzel and K. Sivula, Chem. Sci., 2011, 2, 737 RSC.
- A. Gurlo, S. Lauterbach, G. Miehe, H.-J. Kleebe and R. Riedel, J. Phys. Chem. C, 2008, 112, 9209–9213 CAS.
- X. Li, W. Wei, S. Wang, L. Kuai and B. Geng, Nanoscale, 2011, 3, 718–724 RSC.
- X. Guo, Y. Deng, D. Gu, R. Che and D. Zhao, J. Mater. Chem., 2009, 19, 6706–6712 RSC.
- H. Cao, G. Wang, L. Zhang, Y. Liang, S. Zhang and X. Zhang, ChemPhysChem, 2006, 7, 1897–1901 CrossRef CAS.
- W. Wang, J. Y. Howe and B. Gu, J. Phys. Chem. C, 2008, 112, 9203–9208 CAS.
- H. B. Guo and A. S. Barnard, J. Mater. Chem., 2011, 21, 11566–11577 RSC.
- K. S. Tanwar, C. S. Lo, P. J. Eng, J. G. Catalano, D. A. Walko, G. E. Brown, Jr., G. A. Waychunas, A. M. Chaka and T. P. Trainor, Surf. Sci., 2007, 601, 460–474 CrossRef CAS.
- G. A. Waychunas, Powder Diffr., 2009, 24, 89–93 CrossRef CAS.
- M. A. Henderson, Surf. Sci., 2010, 604, 1197–121 CrossRef CAS.
- D. Spagnoli, B. Gilbert, G. A. Waychunas and J. F. Banfield, Geochim. Cosmochim. Acta, 2009, 73, 4023–4033 CrossRef CAS.
- M. A. Henderson, Surf. Sci., 2002, 515, 253–262 CrossRef CAS.
- F. Jones, A. L. Rohl, J. B. Farrow and W. van Bronswijk, Phys. Chem. Chem. Phys., 2000, 2, 3209–3216 RSC.
- M. A. Henderson, S. A. Joyce and J. R. Rustad, Surf. Sci., 1998, 417, 66–81 CrossRef CAS.
- E. Wasserman, J. R. Rustad, A. R. Felmy, B. P. Hay and J. W. Halley, Surf. Sci., 1997, 385, 217–239 CrossRef CAS.
- E. Wasserman, J. R. Rustad and A. R. Felmy, Surf. Sci., 1999, 424, 19–27 CrossRef CAS.
- J. R. Rustad, E. Wasserman and A. R. Felmy, Surf. Sci., 1999, 424, 28–35 CrossRef CAS.
- S. E. Mason, C. R. Iceman, K. S. Tanwar, T. P. Trainor and A. M. Chaka, J. Phys. Chem. C, 2009, 113, 2159–2170 CAS.
- W. Bergermayer, H. Schweiger and E. Wimmer, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 69, 195409 CrossRef.
- G. J. Martin, R. S. Cutting, D. J. Vaughan and M. C. Warren, Am. Mineral., 2009, 94, 1341–1350 CrossRef CAS.
- A. Rohrbach, J. Hafner and G. Kresse, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 125426 CrossRef.
- H. B. Guo and A. S. Barnard, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 094112 CrossRef.
- J. R. Rustad, Rev. Mineral. Geochem., 2001, 42, 169–198 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter, 1996, 54, 11169 CrossRef CAS.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758 CrossRef CAS.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef.
- S. L. Dudarev, G. A. Botton, S. Y. Savrasov, C. J. Hmphreys and A. P. Sutton, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 1505–1509 CrossRef CAS.
- W. Zhang, J. R. Smith and X.-G. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 024103 CrossRef.
- H. Guo and Y. Qi, Modell. Simul. Mater. Sci. Eng., 2010, 18, 034008 CrossRef.
- J. Majzlan, K.-D. Grevel and A. Navrotsky, Am. Mineral., 2003, 88, 855–859 CAS.
- J. R. Pliego, Jr., Chem. Phys. Lett., 2003, 381, 246–247 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1jm13362d |
| This journal is © The Royal Society of Chemistry 2012 |