The impact of charge defects and resonance enhancement on the two-photon absorption activity of spirofluorene and ladder-type pentaphenylene derivatives†
Namchul
Cho
a,
Gang
Zhou
b,
Kenji
Kamada
c,
Ran Hee
Kim
a,
Koji
Ohta
c,
Sung-Ho
Jin
d,
Klaus
Müllen
b and
Kwang-Sup
Lee
*a
aDepartment of Advanced Materials, Hannam University, Daejeon, 305-811, South Korea. E-mail: kslee@hnu.kr; Fax: +82 42 629 8854; Tel: +82 42 629 8857
bMax-Planck-Institute for Polymer Research, Ackermannweg 10, 55128, Mainz, Germany
cResearch Institute for Ubiquitous Energy Devices, National Institute of Advanced Industrial Science and Technology (AIST), AIST Kansai Center, Ikeda, Osaka 563-8577, Japan
dDepartment of Chemistry Education, Pusan National University, Busan, 609-735, Korea
First published on 20th October 2011
Abstract
Novel two-photon absorption (TPA) chromophores that contain spirofluorene and ladder-type pentaphenylenes (LPPs) as π-centres with diarylamine groups as electron donors (D) in the D–π–D arrangement are prepared. The spirofluorenes with different donors at the both terminals exhibit TPA activity increase as the donor strength increases (from N-ethylcarbazoyl 1, triphenylamino 2, to N,N-dibutylaminophenyl 3). Compared to the spirofluorene derivatives, the LPP derivatives show the larger TPA efficiency due to better coplanarity throughout the molecule. As the LPP unit added from single (4), double (5), to triple (6), the increment of the TPA cross-section exceeds the one-fold of a LPP unit, which can be attributed to the cooperative enhancement of the TPA cross-section (σ2) between the LPP units. We have also explored the effect of introducing charge defects and examined the resonance enhancement by chemical oxidation on the TPA properties of LPP derivatives 4 and 5. Nearly an order of magnitude enhancement was found in the TPA cross-section of dye 4, which was interpreted as the role of charge transfer from LPP π-centre to the cationic diarylamine end groups.
1. Introduction
Two-photon absorption (TPA) in organic molecules has been investigated intensively because of its great potential for use in 3D microfabrication, confocal fluorescence microscopy, photodynamic cancer therapy, and optical-power limiting.1–4 Efficient TPA chromophores are required for all these applications. Thus, substantial efforts have been dedicated to the study of structure–property relationships by means of theoretical and experimental investigations.5–10 In recent times considerable attention has been devoted to enhancing or switching the nonlinear optical responses of chromophores—including the TPA—by means of external stimuli such as pH,11solvent polarity,12 oxidative doping13–15 and electrochemical oxidation.16 The oxidative doping and the electrochemical oxidation of the linear polyenes with organometallic complexes were found to enhance their third-order optical nonlinearities. Theoretical studies clarify that the introduction of charged defects in a π-conjugated system has a marked influence on the TPA activity.5,17,18Ladder-type pentaphenylenes (LPPs) were developed for applications in electroluminescent devices.19,20 These molecules can be considered as linearly condensed fluorene oligomers and therefore exhibit extreme planarity and rigidity at the molecular scale. This is a highly desirable trait for a π-conjugated motif bridging electron withdrawing or donating groups in TPA chromophores. Moreover, oxidized LPPs containing diarylamine groups at both ends of molecules show intervalence charge transfer (IVCT),20 which originates from an optically induced hole transfer between the diarylamine radical-cation of the one end and the neutral diarylamine of the other in the radical monocation species. In addition, another strong electronic transition is observed in the near infrared (NIR) region in dication species. This transition is associated with the charge transfer (CT) from the LPP bridge to the terminal triarylamine groups.21,22 The optical characteristics in these cationic species thus indicate the potential of switching the TPA activity of the LPPs through oxidation.
Here, we report the TPA properties of chromophores that contain LPPs as π-centers with diarylamino substituents as electron donors (D) in the D–π–D molecular arrangement. We investigate three LPP derivatives that differ in the number of LPP repeating units to explore the cooperative enhancement of the TPA cross-section (σ2) between the LPP units. We also investigate chromophores containing vinyl-extended spirofluorene π-centers with different kinds of terminal groups (i.e., N-ethylcarbazoyl 1, triphenylamino 2, and N,N-dibutylaminophenyl 3) and compare their TPA properties with those of an LPP derivative of similar size. In addition, we examine the impact of introducing charge defects and the effect of the resonance enhancement by chemical oxidation on the TPA properties of one of the LPP derivatives, in what could be the first example of LPP-based cationic TPA chromophores.
2. Results and discussion
2.1 One-photon absorption and fluorescence
As shown in Scheme 1, two different series of two-photon absorbing donor–π–donor (D–π–D) type chromophores are prepared. One of the series has a vinyl-extended spirofluorene unit as the π-conjugation center, with electron donors of different strengths. The other series has ladder type pentaphenylene (LPP) oligomer groups as the π-conjugation center, with diarylamine units at the end of molecules. Spirofluorene derivatives 1–3 were synthesized by the Heck coupling reaction of vinyl-substituted 9-ethylcarbazole, triphenylamine, and N,N-dibuthylaniline with 2′,7′-dibromo-3,6-bis(3,7-dimethyloctyloxy)-9,9′-spirobifluorene. LPP derivatives 4–6 were prepared following a procedure reported in the literature.20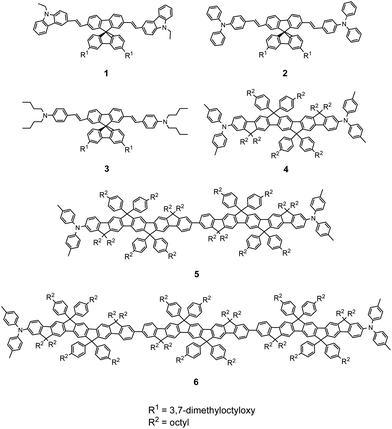 | ||
| Scheme 1 Molecular structures for TPA compounds 1–6. | ||
Spirofluorene derivatives 1–3 have broad, intense, one-photon absorption bands with substructures around 360–450 nm and intense one-photon-induced fluorescence bands around 420–550 nm (Fig. 1 and Table 1). The absorption and fluorescence maxima (λabs(1) and λflu(1)) were red shifted upon changing the terminal groups from N-ethylcarbazole to triphenylamine, and to N,N-dibutylaminophenyl. A similar red shift was previously reported for dithienothiophene compounds containing the same terminal donors (N-ethylcarbazole and triphenylamine) in a donor–π–donor (D–π–D) arrangement.23 The fluorescence quantum yield (Φf) for compounds 1–3 decreases with increasing donor strength. The transition dipole moments (Mge: 11.7–12.9 D) are similar for all three compounds (Table 2). Using the radiative lifetime, the fluorescence quantum yield (Φf) can also be written as Φf = τs/τr, where τs is the lifetime of excited state and τr is the radiative lifetime. The radiative (krad) and non-radiative decay rates (knrad) can be calculated by relations as krad = Φf/τs and knrad = (1/τs)(1 − Φf). Fluorescence-lifetime measurements performed using time-correlated single-photon counting (TCSPC) suggest that the decrease in Φf originates from an increase in the knrad, which is caused by the increased solute–solvent interactions with the strong terminal donors, and a decrease in the krad (Table 1).
 | ||
| Fig. 1 (a) One-photon absorption and (b) fluorescence spectra for compounds 1–6. | ||
| TPA dyes | λ abs (1)/nm | ε b/103 M−1 cm−1 | λ flu (1)/nm | Δνc | Φ f d | τ e/ns (in toluene) | k rad f/108 s−1 | k nrad f/108 s−1 |
|---|---|---|---|---|---|---|---|---|
| a Absorption and emission spectra were obtained in THF. b Molar extinction coefficient. c Stokes shift. d Fluorescence quantum yield. e Fluorescence lifetime measured by time-correlated single-photon counting (TCSPC). f Rate constants for radiative (krad) and non-radiative (knrad) decays. | ||||||||
| 1 | 398 | 110 | 439 | 2347 | 0.87 | 0.94 | 9.26 | 1.38 |
| 2 | 410 | 133 | 457 | 2508 | 0.71 | 1.07 | 6.64 | 2.71 |
| 3 | 416 | 109 | 493 | 3754 | 0.63 | 1.11 | 5.68 | 3.33 |
| 4 | 429 | 103 | 452 | 1186 | 0.83 | 0.88 | 9.43 | 1.93 |
| 5 | 432 | 249 | 453 | 1314 | 0.78 | 0.73 | 10.68 | 3.01 |
| 6 | 433 | 239 | 454 | 1068 | 0.80 | 0.64 | 12.50 | 3.13 |
| TPA dyes | E ge a/eV | E ge′ b/eV | ΔEc/eV | M ge d/D | Mee′e/D | λ max(2) f/nm | σ 2 g/GM | σ 2 h/N |
|---|---|---|---|---|---|---|---|---|
| a Transition energy between the ground state and the one-photon-allowed excited state. b Transition energy between the ground state and the two-photon-allowed excited state. c Detuning energy ΔE = Ege − 1/2Ege′. d Transition dipole moment calculated from eqn (2) in the Experimental section. e Transition dipole moment calculated from the first relation of eqn (2). f Maximum of the two-photon fluorescence excitation spectrum. g Two-photon absorption cross-section (1 GM = 1 × 10−50 cm4 s photon−1 molecule−1). h N is the number of double bonds in compounds (the phenyl rings are considered to be 1.5 double bonds). Experimental uncertainty: ±15%. i Measured by the Z-scan method because of the limitations of the two-photon-induced fluorescence setup at wavelengths below 700 nm (Fig. 2). | ||||||||
| 1 | 3.12 | — | — | 11.9 | — | — | 400 | 36 |
| 2 | 3.02 | 3.31 | 1.37 | 12.9 | 14.4 | 750 | 530 | 66 |
| 3 | 2.98 | 3.31 | 1.33 | 11.7 | 20.6 | 750 | 950 | 119 |
| 4 | 2.89 | 3.67 | 1.06 | 9.2 | 18.7 | 676 | 940i | 125 |
| 4 2+ | 1.01 | 2.75 | 0.36 | 28.4 | 10.1 | 899 | 8900i | 1187 |
| 5 | 2.87 | 3.49 | 1.12 | 13.6 | 21.3 | 710 | 2120 | 141 |
| 5 2+ | 0.94 | 2.67 | 0.40 | 16.3 | 28.0 | 930 | 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 300i 300i |
1154 |
| 6 | 2.86 | 3.49 | 1.12 | 13.1 | 27.8 | 710 | 3400 | 191 |
Compared to compounds 1 and 2, compound 3 exhibits a broader fluorescent band with a large Stokes shift (Δν), thus suggesting a large conformational change in its relaxed excited state due to strong interactions with the environment.
All LPP derivatives 4, 5 and 6 having single, double and triple repeating units of LPP exhibit a sharp, intense, one-photon absorption band with prominent vibronic structures around 430 nm (Fig. 1a). Both spectral parameters, λabs(1) and λflu(1), show a red shift as increasing the number of repeating LPP units; however, these shifts are very small. These small red shifts suggest that the effective conjugation length is limited within the LPP single unit (Table 1). Hence, the values of the Stokes shift are not much different for compounds 4–6. In addition, all LPP compounds exhibit a high fluorescence quantum yield (Φf = 0.78–0.83), with very similar values. These spectroscopic observations suggest that the geometrical changes are hardly found in the LPP series between the ground and excited states, as expected from the rigidity of the LPP unit.19,20
2.2. Two-photon absorption
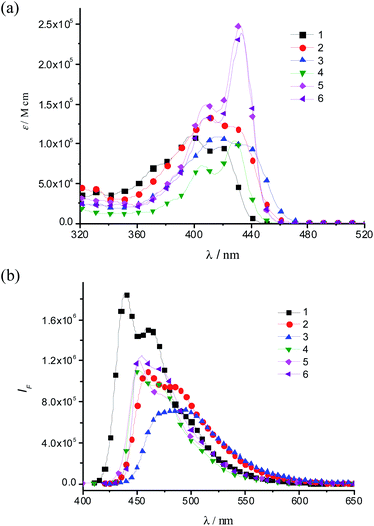
Fig. 2a shows the two-photon-induced fluorescence (TPIF) excitation spectra of the spirofluorene compounds 1–3 obtained from femtosecond (fs) TPIF measurements. Both compounds of 2 and 3 show TPA maxima at the same wavelength (750 nm), but the peak TPA cross-section σ2 for 3 (950 GM) is much higher than that for 2 (530 GM). On the other hand, the spectral amplitude of 1 is smaller than those of the other two. The TPA peak position of 1 is probably located at a shorter wavelength, beyond the range of our measurement setup; therefore, we excluded this compound from the discussion here even the observed σ2 value of 3 is 400 GM at the wavelength of 700 nm. This result indicates that the donor strength plays a crucial role in determining the TPA intensity.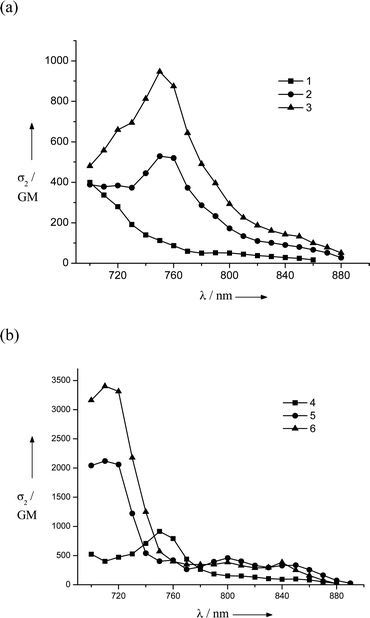 | ||
| Fig. 2 Two-photon-induced fluorescence excitation spectra for compounds (a) 1–3 and (b) 4–6. | ||
The TPA cross-section can be represented as a function of the photon energy of the incident light (Eph) by using the three-state model based on the sum-over-states (SOS) expression (eqn (1)):
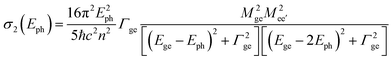 | (1) |
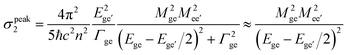 | (2) |
It has been reported that strong donors accelerate the symmetric intramolecular charge transfer (ICT), thus resulting in large TPA activity through a large breaking of the alternant symmetry, which can cause an enhancement of the transition dipole moment between the excited states (Mee′).5,9 Spectroscopic parameters are summarized in Table 2. The larger Mee′ value (20.6 D) and the smaller detuning energy (1.33 eV) for compound 3 compared to those of compound 2 (14.4 D and 1.37 eV, respectively) contribute to the larger TPA cross-section of 3. Therefore, the origin of the increasing TPA cross-sections can be explained by the increasing Mee′ values that result from the large breaking of the alternant symmetry caused by the introduction of strong terminal donors.
Comparison between compounds 4 and 2 (which have similar terminal groups) clarifies the impact of the nature of the π-centers on the spectroscopic properties. The one-photon absorption peak of 4 is located at a longer wavelength and has a narrower bandwidth compared to that of 2 (Fig. 1a). This red shift indicates that the LPP π-center provides a more efficient electronic communication than the spirofluorene π-center. On the other hand, the narrower bandwidth suggests that the interactions of 4 with the surroundings are weaker than those of 2. Moreover, the Stokes shift of 4 is much smaller (almost the half) than that of 2 (Table 1). These facts can be also interpreted by considering the difference in structural rigidity between the LPP and spirofluorene π-centers.
The TPA cross-section for compound 4 was evaluated by the Z-scan method because of the limitations of the TPIF setup at wavelengths below 700 nm. The observed TPA cross-section value (σ2) of 4 shows 940 GM at the wavelength of 676 nm. This σ2 value is almost two times that of 2 (530 GM). This enhancement is due to a longer conjugation pathway and the rigid planar structure of the LPP unit, which facilitate π-conjugation over the molecule. Moreover, these peak σ2 values are much larger than that reported previously for smaller fluorene derivatives containing the same electron donors as 4 and 2 [namely, 9,9-didecyl-2,7-bis(N,N-diphenylamino)fluorene, which has a peak σ2 ≈ 100–240 GM in polar and nonpolar solvents].26 These facts clearly show that an appropriate selection of the π-centers has a significant impact on the TPA efficiency. The comparison between the reported fluorene derivative [9,9-didecyl-2,7-bis(N,N-diphenylamino)fluorene] and the extended compound 4 shows that a longer conjugation pathway of the π-center enhances the TPA efficiency. This enhancement is further boosted by the rigid planar structure of the LPP unit, which provides a more intense conjugation over the π-center.
As shown in eqn (2), at TPA resonance, three factors can contribute to a large TPA cross-section, namely, a small ΔE, a large Mge, and a large Mee′. Compound 4 has a smaller Mge (9.2 D) than 2 (12.9 D). However, it has a smaller ΔE (1.06 eV for 4, 1.37 eV for 2) and a larger Mee′ (18.7 D for 4, 14.4 D for 2), which contribute to the larger σ2 of 4. The above discussions are also supported by geometry-optimization calculations. To perform such calculations, we constructed simplified structures (2-1 and 4-1) by replacing all the peripheral octyl and terminal phenyl groups linked to the amine centers of 2 and 4 with methyl groups. In the optimized ground-state geometries in AM1 level as shown in Fig. 3, chromophore 4-1 shows a more planar structure relative to 2-1.
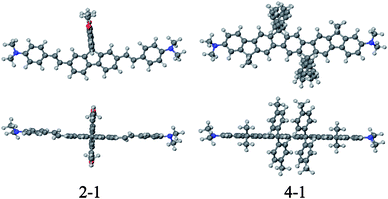 | ||
| Fig. 3 Ground-state optimized geometries of simplified molecules at the AM1 level, 2-1 and 4-1: top (bottom) and side (top) views relative to the molecular plane. | ||
In the TPA spectra of LPP derivatives 4–6, all compounds exhibit very strong TPA bands (Fig. 2b), and the peak TPA cross-section significantly increases with an increase in the number of LPP repeating units (σ2 = 940, 2120 and 3400 GM for 4, 5 and 6, respectively), while the one-photon absorption cross-section (∝ε) saturates with the extension of the LPP unit. The TPA cross-sections of LPP derivatives 5 and 6 are, respectively, 2.3 and 3.7 times larger than that of 4. The large Mee′ values for compounds 5 and 6 are probably responsible for the large increase in the TPA activity, even though there are slight increases in the detuning energy ΔE (Table 2). It is also worth noting that the Mge value does not increase between 5 and 6 (because there is no electronic communication between two arylamine moieties and each LPP moiety behaves independently20), whereas the TPA cross-section does increase cooperatively. This nonlinear increase in the TPA cross-section for compounds 4–6 suggests that the LPP repeating units are coupled to each other through nonlinear absorption. It has been reported that polyfluorene displays a larger enhancement in the TPA cross-section values in the visible range compared to bifluorene.27 Investigations of the effect of increasing the number of LPP repeating units showed that dipolar coupling between the LPP units leads to an increase in the TPA activity and a red shift of the absorption band. On the other hand, the stronger nonlinearities of the TPA activity of dendritic and/or multibranched TPA chromophores compared to dipolar and quadrupolar chromophores result from cooperative effects related to a sizeable electronic coupling between branches.28,29 Accordingly, we can conclude that the nonlinear increase in the TPA cross-section of 6 originates from the cooperative coupling between LPP repeating units. Therefore, the cooperative interaction between LPP units can provide a way to design TPA chromophores with high efficiency and opens up the possibility of obtaining outstanding nonlinear optical materials in the near infrared (NIR) regime.
2.3. The impact of charge defect on the TPA properties of the LPP compounds
To investigate the impact of charge defect on TPA properties we generated cations by oxidizing LPP derivative 4 with SbCl5. Depending on the amount of SbCl5 the monocation or dication can be generated. When 1.5 equiv. of SbCl5 is employed with respect to 1 equivalent (equiv.) of 4 monocation is generated, whereas 3.0 equiv. of SbCl5 prominently gives rise to dication. The evolution of the UV-Vis-NIR absorption spectrum of a solution of 4 in toluene upon addition of different equiv. of SbCl5 is shown in Fig. 4. The evolution shows the same trend as that reported previously for a solution of the same compound in dichloromethane.20 Upon increasing the equiv. of SbCl5, the intense band at 429 nm (23148 cm−1, assigned to the HOMO–LUMO transition) disappears while new bands appear around 450 nm (22222 cm−1) and 528 nm (18148 cm−1) at 1.6 equiv. of SbCl5. These bands are assigned to the diphenylamine cation and are probably due to the SOMO–LUMO and HDOMO–SOMO transitions, respectively (SOMO = singly occupied molecular orbital; HDOMO = highest doubly occupied molecular orbital). These bands more or less blue-shift at SbCl5 concentrations of 2 equiv. or more, thus suggesting a change of the SOMO level into the dicationic form. In the NIR region, a new band appears at 1510 nm (6222 cm−1) at 1.2 equiv. of SbCl5. At 1.6 equiv. or more, this band disappears and another intense one, centered at 1232 nm (8117 cm−1), appears. The band at 1510 nm can be described as the IVCT band of the cation radical (4˙+), whereas that at 1232 nm can be assigned to CT processes from the π-center to the triphenylamine cation centers of the dication (42+). These bands are blue-shifted compared to those reported in dichloromethane (5283 and 7184 cm−1, respectively).20 These blue shifts are probably due to the less-polar environment in toluene compared to dichloromethane and support the assignment of these bands to CT processes. A small shoulder always appeared at 1018 nm (9823 cm−1), irrespective of the SbCl5 concentration. This small signal can be assigned to the CT band because it shows a blue shift compared to the signal in dichloromethane (8850 cm−1). A similar core-to-terminal CT band was also found in the case of another bis(triphenylamine) compound with a different π-center.21,22
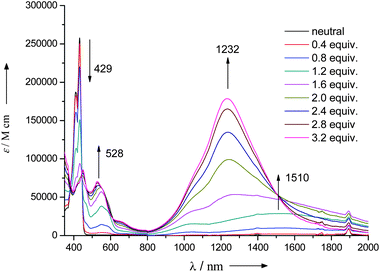 | ||
| Fig. 4 UV-Vis-NIR absorption spectra of compound 4 in toluene solutions upon stepwise addition of SbCl5. | ||
TPA spectrum of a solution of 4 (ca. 5 × 10−4 M) in toluene (at 3.2 equiv. of SbCl5) was obtained using the femtosecond open-aperture Z-scan method. Compound 4 was mostly oxidized and converted into dication 42+ (at 3.2 equiv. of SbCl5). As a control experiment, we also measured the spectrum of a solution of 4 without SbCl5 under the same conditions. Fig. 5 shows the obtained TPA spectra for the solutions of 4 with and without SbCl5. The obtained TPA spectrum of 4 by the Z-scan method is consistent with that by the TPIF method (Fig. 2b). It was found that the oxidation of 4 drastically enhances its TPA activity. The peak σ2 value for the oxidized form of this compound reaches 8900 GM at 899 nm (a conversion to 42+ of almost 100% is assumed), which is nearly one order of magnitude larger than that of neutral 4 (940 GM at 676 nm).30 The large red shift of the TPA peak of oxidized 4 suggests that this very intense band must originate from the dication 42+, which is the dominant species at this SbCl5 concentration. The magnitude of Mge for 42+ increases significantly with respect to the neutral compound 4 (from 9.2 to 28.4 D) because of the increased CT band at 1232 nm (Table 2). This result implies that the introduction of charged defects facilitates π-electron delocalization over the entire molecule and, consequently, causes an enhancement of the TPA cross-section. Since the one-photon absorption spectrum of the dication 42+ shows a characteristic, intense CT band centered at 1232 nm, resonance enhancement through this intense CT band could also be a plausible mechanism for explaining the drastic enhancement observed.5,10 Similar studies were conducted with the dication 52+, which also showed a drastic enhancement in TPA characteristics (17300 GM).
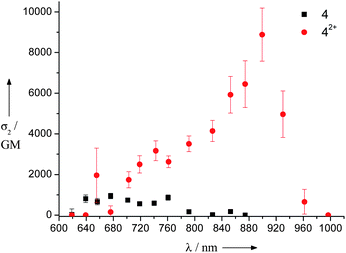 | ||
| Fig. 5 Two-photon absorption spectra of LPP derivative 4 in toluene with 3.2 equiv. of SbCl5 (circles) and without SbCl5 (squares) measured using the Z-scan method. | ||
To examine the effect of CT, we calculated the enhancement factor at the TPA peak (defined as F ≡ Mge2/ΔE2) for each one-photon absorption band of 42+ and tried to determine which one-photon absorption state plays the most important role as intermediate state in the enhancement process. We found that the bands at 1232 and 1018 nm have much larger F values than other bands, which suggests that these two one-photon absorption bands are the main contributions to the resonance enhancement of the TPA band (Table 3). The bands at 1232 and 1018 nm originate from a CT process from the π-center to the triphenylamine cation centers; therefore, we can conclude that the CT states act as the dominant intermediate states for the TPA transition, thus giving rise to the drastic enhancement. This discussion of the resonance enhancement is based only on the detuning term which appears in the denominators of eqn (1) and (2). These results show that the introduction of charge defects like dications is an alternative route to design efficient TPA materials and modify the structure of TPA molecules. The dications can be generated not only by chemical oxidation but also by electrochemical means. These results suggest a new possibility of switching the properties of TPA through its oxidation state, as has been reported before for Ru complexes.16 It is also worthy to note here that the magnitude relation between the transition moments is counterchanged by the oxidation; i.e. Mge < Mee′ for 4 while Mge > Mee′ for 42+ (Table 2 and Fig. 6). This can be explained by difference in the nature of the electronic transitions between the neutral and dicationic states. For the neutral state, the g–e transition is dominated by the weaker HOMO–LUMO transition and the e–e′ transition is governed by the combination of the stronger HOMO–HOMO − 1 and LUMO–LUMO + 1 transitions.5 On the other hand, for the dicationic state, where two electrons are removed from HOMO and then HOMO − 1 becomes the new highest occupied frontier orbital accompanying change of the bonding structure to the quinoidal, the stronger HOMO–HOMO − 1 transition is now the main contribution to the lowest-energy, i.e. g–e, transition and the weaker HOMO–LUMO transition governs the e–e′ transition. That is, the roles of the HOMO–LUMO and HOMO–HOMO − 1 transitions to the g–e and e–e′ transitions are counterchanged and therefore the magnitude relations are also counterchanged.
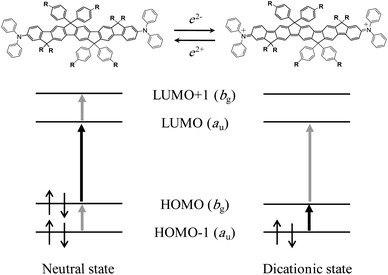 | ||
| Fig. 6 Molecular orbital diagram of the neutral and dicationic states of LPP derivative 4. The black and gray thick arrows illustrate the proposed dominant contributions to the g–e and e–e′ transitions, respectively. Shift of the energy level of the orbitals in the dicationic state as observed for the blue shift of the HDOMO–SONO transition in Fig. 4 (see text) is ignored for schematic simplicity in order to show the correlation of the orbitals between the neutral and dicationic states. | ||
3. Conclusions
We have studied the TPA activities of a new series of two-photon absorbing molecules based on spirofluorene and LPP. Especially LPP derivatives exhibited a considerably large TPA cross-section (940–3400 GM). The TPA activity enhancement for compounds 4–6 can be explained by a cooperative coupling between the LPP building blocks. In addition, the magnitude of σ2 was not saturated—even with a very long conjugation for 6—so we can expect a much larger σ2 value by increasing the conjugation length in our LPP series, for example, by using higher repeating units of LPP than the reported materials in our work. Moreover, we have investigated the effect of charge defect, introduced by chemical oxidation, on the TPA activity. We found that resonance enhancement by the intense CT band due to charge transfer from the LPP π-center to the triarylamine cations induces the strong TPA transition.Acknowledgements
This work was supported by the IRTG program and the Active Polymer Center for Patterned Integration (ERC R11-2007-050-01002-0) of the National Research Foundation of Korea (NRF), German Research Foundation (DFG), and KAKENHI (Grant-in-Aid for Scientific Research) on Priority Area “Strong Photon-Molecule Coupling Fields (No. 470)” from the Ministry of Education, Culture, Sports, Science and Technology of Japan. The authors thank Prof. P. L. Baldeck at Université Joseph Fourier for the help with the TPIF measurement, and also Prof. D. Kim at the Yonsei University for the help with the fluorescence lifetime measurement.References
- (a) B. H. Cumpston, S. P. Ananthavel, S. Barlow, D. L. Dyer, J. E. Ehrlich, L. L. Erskine, A. A. Heikal, S. M. Kuebler, I.-Y. S. Lee, D. McCord-Maughon, J. Qin, H. Röckel, M. Rumi, X. Wu, S. R. Marder and J. W. Perry, Nature, 1999, 398, 51 CrossRef CAS; (b) H. Sun, T. Kawakami, Y. Xu, J. Ye, S. Matuso, H. Misawa, M. Miwa and R. Kaneko, Opt. Lett., 2000, 25, 1110 CrossRef CAS; (c) K.-S. Lee, R. H. Kim, D.-Y. Yang and S. H. Park, Prog. Polym. Sci., 2008, 33, 631 CrossRef CAS; (d) S. H. Park, T. W. Lim, D.-Y. Yang, N. C. Cho and K.-S. Lee, Appl. Phys. Lett., 2006, 89, 173133 CrossRef.
- (a) A. Abbotto, L. Beverina, R. Bozio, S. Bradamante, C. Ferrante, G. A. Pagani and R. Signorini, Adv. Mater., 2000, 12, 1963 CrossRef CAS; (b) J. H. Strickler and W. W. Webb, Opt. Commun., 1991, 16, 1780 CAS; (c) H. M. Kim and B. R. Cho, Acc. Chem. Res., 2009, 42, 863 CrossRef CAS; (d) H. M. Kim and B. R. Cho, Chem.–Asian J., 2011, 6, 58 CrossRef CAS; (e) S. Sumalekshmy and C. J. Fahrni, Chem. Mater., 2011, 23, 483 CrossRef CAS.
- (a) W. R. Dichtel, J. M. Serin, C. Edder, J. M. J. Fréchet, M. Matuszewski, L. S. Tan, T. Y. Ohulchansky and P. N. Prasad, J. Am. Chem. Soc., 2004, 126, 5380 CrossRef CAS; (b) G. S. He, P. P. Markowicz, T. C. Lin and P. N. Prasad, Nature, 2002, 415, 767 CAS.
- (a) J. B. Ehrlich, X. Wu, I.-Y. S. Lee, Z. Y. Hu, H. Rockel and S. R. Marder, Opt. Lett., 1997, 22, 1843 CrossRef CAS; (b) D. C. Neckers, W. S. Jenks and T. Wolff, Adv. Photochem., 2007, 29, 111 Search PubMed; (c) G. S. He, L.-S. Tan, Q. Zheng and P. N. Prasad, Chem. Rev., 2008, 108, 1245 CrossRef CAS; (d) H. M. Kim and B. R. Cho, Chem. Commun., 2009, 153–164, 10.1039/B813280A.
- K. Ohta and K. Kamada, J. Chem. Phys., 2006, 124, 124303 CrossRef.
- M. Albota, D. Beljonne, J.-L. Brédas, J. E. Ehrlich, J. Y. Fu, A. A. Heikal, S. E. Hess, T. Kogej, M. D. Levin, S. R. Marder, D. McCord-Maughon, J. W. Perry, H. Röckel, M. Rumi, G. Subramaniam, W. W. Webb, X.-L. Wu and C. Xu, Science, 1998, 281, 1653 CrossRef CAS.
- D. Beljonne, Z. Shuai, J. Cornil, D. A. Dos Santos and J.-L. Brédas, J. Chem. Phys., 1999, 111, 2829 CrossRef CAS.
- M. Drobizhev, Y. Stepanenko, Y. Dzenis, A. Karotki, A. Rebane, P. N. Taylor and H. L. Anderson, J. Phys. Chem. B, 2005, 109, 7223 CrossRef CAS.
- M. Rumi, J. E. Ehrlich, A. A. Heikal, J. W. Perry, S. Barlow, Z. Hu, D. McCord-Maughon, T. C. Parker, H. Rockel and S. Thayumanavan, J. Am. Chem. Soc., 2000, 122, 9500 CrossRef CAS.
- K. Kamada, K. Ohta, Y. Iwase and K. Kondo, Chem. Phys. Lett., 2003, 372, 386 CrossRef CAS.
- M. H. V. Werts, S. Gmouh, O. Mongin, T. Pons and M. Blanchard-Desce, J. Am. Chem. Soc., 2004, 126, 16294 CrossRef CAS.
- B. Strehmel, A. M. Sarker and H. Detert, ChemPhysChem, 2003, 4, 101 CrossRef.
- K. Meerholz, J. Swiatkiewicz and P. N. Prasad, J. Phys. Chem., 1995, 99, 7715 CrossRef CAS.
- C. W. Spangler and M. He, J. Chem. Soc., Perkin Trans. 1, 1995, 715 RSC.
- C. W. Spangler, P. K. Liu and K. O. Havelka, J. Chem. Soc., Perkin Trans. 2, 1992, 1207 RSC.
- M. Cifuentes, C. Powell, M. Humphrey, G. Heath, M. Samoc and B. Luther-Davies, J. Phys. Chem. A, 2001, 105, 9625 CrossRef CAS.
- H. Fujita, M. Nakano, M. Takahata and K. Yamaguchi, Chem. Phys. Lett., 2002, 358, 435 CrossRef CAS.
- R. Kishi, M. Nakano, S. Yamada, K. Kamada, K. Ohta, T. Nitta and K. Yamaguchi, Chem. Phys. Lett., 2004, 393, 437 CrossRef CAS.
- J. Jacob, S. Sax, T. Piok, E. J. W. List, A. C. Grimsdale and K. Müllen, J. Am. Chem. Soc., 2004, 126, 6987 CrossRef CAS.
- G. Zhou, M. Baumgarten and K. Müllen, J. Am. Chem. Soc., 2007, 129, 12211 CrossRef CAS.
- C. Lambert and G. Nöll, J. Chem. Soc., Perkin Trans. 2, 2002, 2039 RSC.
- C. Lambert, G. Nöll and J. Schelter, Nat. Mater., 2002, 1, 69 CrossRef CAS.
- O. K. Kim, K.-S. Lee, H. Woo, K. S. Kim, G. S. He, J. Swiatkiewicz and P. N. Prasad, Chem. Mater., 2000, 12, 284 CrossRef CAS.
- B. J. Orr and J. F. Ward, Mol. Phys., 1971, 20, 513 CrossRef CAS.
- Y. R. Shen, Principles of Nonlinear Optics, Wiley-Interscience, NewYork, USA, 1984 Search PubMed.
- K. D. Belfield, A. R. Morales, J. M. Hales, D. J. Hagen, E. W. Van Stryland, V. M. Chapela and J. Percino, Chem. Mater., 2004, 16, 2267 CrossRef CAS.
- P. Najechalski, Y. Morel, O. Stephan and P. L. Baldeck, Chem. Phys. Lett., 2001, 343, 44 CrossRef CAS.
- W. J. Yang, D. Y. Kim, C. H. Kim, M. Y. Jeong, S. K. Lee, S. J. Jeon and B. R. Cho, Org. Lett., 2004, 6, 1389 CrossRef CAS.
- C. Katan, F. Terenziani, O. Mongin, M. H. V. Werts, L. Porres, T. Pons, J. Mertz, S. Tretiak and M. Blanchard-Desce, J. Phys. Chem. A, 2005, 109, 3024 CrossRef CAS.
- (a) L. Porres, V. Alain, L. Thouin, P. Hapiot and M. Blanchard-Desce, Phys. Chem. Chem. Phys., 2003, 5, 4576 RSC; (b) R. Rathore, A. S. Kumar, S. V. Lindeman and J. K. Kochi, J. Org. Chem., 1998, 63, 5847 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1jm13481g |
| This journal is © The Royal Society of Chemistry 2012 |