Tailored star-shaped statistical teroligomers viaATRP for lithographic applications
Florian
Wieberger†
a,
Drew C.
Forman†
b,
Christian
Neuber
a,
André H.
Gröschel
c,
Marietta
Böhm
c,
Axel H. E.
Müller
c,
Hans-Werner
Schmidt
*a and
Christopher K.
Ober
*b
aMacromolecular Chemistry I, University of Bayreuth, Universitätstraβe 30, 95440, Bayreuth, Germany
bMaterials Science & Engineering, Cornell University, 210 Bard Hall, Ithaca, NY 14853, USA
cMacromolecular Chemistry II, University of Bayreuth, Universitätstraβe 30, 95540, Bayreuth, Germany
First published on 14th October 2011
Abstract
A series of five star-shaped teroligomers consisting of a saccharose core, and arms, composed of α-gamma butyrolactone methacrylate (GBLMA), methyl adamantyl methacrylate (MAMA) and hydroxyl adamantyl methacrylate (HAMA) with defined arm length and number of arms were prepared via the core-first atom transfer radical polymerization (ATRP) route. The saccharose core was modified with ATRP initiating sites and non-reactive sites, enabling the synthesis of star polymers with a smaller arm number but identical core. Star teroligomers were synthesized with narrow molecular weight distributions with low polydispersity indices (PDIs < 1.1) showing negligible side reactions only at higher conversions of Xp > 50%. The absence of side reaction and the precise achievement of the target molecular weight indicated excellent control over the reaction. A selected star-shaped teroligomer was investigated for the first time as a photoresist material. The delicate conditions of the lithographic process were optimized by a combinatorial approach. The obtained low line edge and line width roughness of the observed pattern demonstrate the potential of the star architecture for this application.
Introduction
Star-shaped polymers, which may consist of multiple linear homo-, block, or statistical copolymer arms covalently attached to a central core, have attracted interest in numerous research fields due to properties that can be tuned by varying the number, length and chemical nature of the arms.1–5 Star-shaped polymers with very few arms behave in a similar fashion to their linear counterparts. However, as the number of arms increases, their ability to interact with neighboring stars decreases, in essence trading intermolecular for intramolecular interactions, leading to their description as “ultrasoft colloids”.3 In this regime, star-shaped polymers possess properties significantly different from their linear counterparts. As a result stars are used for a range of applications including viscosity index modifiers in oil,6oxygen permeability and hardness enhancers in contact lenses,7 additives in coatings, binder in toner and as an encapsulation material for pharmaceuticals.8,9 The star-shaped polymer architecture continues to be the focus of both theoretical and experimental studies, which explore the relationship between architecture and thermodynamics, osmotic pressure, molecular size, rheology and polyelectrolyte behavior.10–15There are many areas where the properties of the star-shaped architecture could prove beneficial, such as conjugates for drug delivery,16nanoparticle dispersion stabilizers,17 cosmetic dyes,18 and in vivo sensors.19 With this work star-shaped polymers are introduced to the research field of lithography as new photoresist materials. Standard resists are still based on linear polymer materials.20 During the last decade an additional architecture, molecular glasses (MG), introduced by Shirota et al.,21 has been studied as a new class of photoresist materials.22,23 The main advantages of MGs are the formation of transparent films, their monodispersity, the usage of common organic purification methods and the smaller molecule size, which is about the same size as additives such as a photo acid generator and base quencher.24 The advantages of common polymer materials are stable amorphous film formation, robust processability and the almost unlimited structural flexibility due to monomer choice and architecture.22 In contrast to MGs, polymers show chain entanglements and have an average coil dimension of 5–10 nm. Chain entanglements and extended coil dimension are considered to be the main reasons for irregularities in high-resolution patterns. The introduction of star-shaped polymer architecture is intended to overcome issues from both the linear polymer and the MG photoresist materials. Additional research in non-linear polymer architectures has included graft25 or hyperbranched26 polymer resists that were designed to improve the resist's dissolution behavior or to enhance the sensitivity of the resist, respectively. These works demonstrate that switching from linear polymers to more complex architectures can yield improved performance.
Anionic polymerization is a well-known and well-established method for the fabrication of tailor made polymers with precise control over molecular weight (MW) and (low) polydispersities.27 However, this method exhibits major drawbacks in modern material research as it is highly susceptible to impurities (especially oxygen and water) as well as functional groups (electrophilic, polar and protic). Although new synthetic paths like RAFT and NMP overcome some of these obstacles, they involve long and difficult preparative efforts and complicated synthetic paths towards star-shaped polymers.28 In the past two decades, atom transfer radical polymerization (ATRP) has emerged as one of the most versatile controlled radical polymerization techniques sustaining most functional groups and even small amounts of oxygen.29 In addition, reaction conditions can be easily tuned by many parameters for accurate polymer synthesis. Another interesting feature is the straightforward preparation of multifunctional, brush- or even star-shaped initiators by simple and inexpensive one-step esterification of oligo- and polyols with halogen bearing moieties.30,31 The possibility of creating architectures for a wide range of applications is almost infinite as the pool of monomers polymerizable viaATRP is still increasing.28 When ATRP is used to prepare copolymers different conversions yield different compositions except for the case of monomer feeds with the same reactivity.32
Based on this knowledge, we address key challenges facing ATRP synthesis of star-shaped statistical teroligomers. While conversion was previously utilized as a process variable, in this study it will be kept in a narrow range, while the reaction rate is controlled by the [CuI]/[CuII] and [M]:[I] ratios. This allows the synthesis of stars comprised of different cores with different arm lengths to be prepared at similar conversions and hence, identical composition. Introducing specific fractions of non-reactive sites into the saccharose initiator proved successful in controlling the average arm number of synthesized star teroligomers. With reaction conditions and polymerization kinetics resolved one specific well-defined star architecture was used to investigate its potential in lithographic patterning.
Experimental part
Materials
Ethyl 2-bromoisobutyrate (EBiB) (97%), 2-bromoisobutyryl bromide (98%), propionyl bromide (98%), 4-(dimethylamino)pyridine (99%), pyridine, dichloromethane, copper(I) chloride (>99%), copper(II) chloride (>99%), and N,N,N′,N′′,N′′- pentamethyldiethylenetriamine (PMDETA) (98%), triphenylsulfonium perfluoro-1-butanesulfonate (>99%), propylene glycol monomethyl ether acetate (99%), tetramethylammonium hydroxide solution (10 wt.%), hexamethyldisilazane (HMDS) were purchased from Aldrich and D(+)-saccharose was purchased from Aldi Süd. Methyl adamantyl methacrylate (MAMA) and hydroxyl adamantyl methacrylate (HAMA) were purchased from Idemitsu Chemicals and α-gamma butyrolactone methacrylate (GBLMA) from Kuraray. MAMA and GBLMA were passed through a basic alumina column to remove the inhibitor. HAMA was used as received. All solvents used were of analytical grade.Preparation of multifunctional initiators
The core-first33 approach and ATRP were selected to synthesize star-shaped oligomers. 2,3,4,6,1′,3′,4′,6′-octa-O-(2-bromoisobutyryl)-saccharose (1), which has eight initiating sites, was prepared from saccharose as previously published.34A modified multifunctional initiator with fewer initiating sites (2) was prepared by substituting the 2-bromoisobutyryl bromide with a 50/50 solution of 2-bromoisobutyryl bromide and propionyl bromide. 5.0 g (14.6 mmol) D(+)-saccharose was dehydrated in a vacuum oven at 80 °C for 1 h prior to being stirred with 4-(dimethylamino)pyridine in catalytic quantities in a solution of 200 mL chloroform and 90 mL pyridine under nitrogen. After cooling the solution with ice, 26.9 g (116.8 mmol) 2-bromoisobutyryl bromide and 16.0 g (116.8 mmol) propionyl bromide were added from a dropping funnel over a period of 3 h. The solution was brought to room temperature and stirred overnight before undergoing reflux at 75 °C for three hours. The solution was diluted with diethyl ether and the pyridinium bromide was removed by repeated extraction with water, sodium hydrogen carbonate solution and sodium chloride solution. The solution was concentrated and purified by column chromatography (SiO2; ethyl acetate), the solvent evaporated, then dissolved in hot methanol and precipitated at −20 °C over night. The obtained solid mixture of products corresponding to the saccharose based multifunctional initiator 2 has an average of 3.5 initiating sites. (6.2 g, 5.6 mmol, 38.1%)
Eight site saccharose-based initiator 1: M (+Li+) = 1.541 g mol−1 by MALDI-TOF MS (DHB:LiCl:initiator 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1).34 Reduced site saccharose-based initiator 2: M (+Li+) = 889 (5.8%), 981 (17.6%), 1.073 (27.6%), 1.165 (25.8%), 1.257 (14.5%), 1.348 (8.7%) g mol−1 by MALDI-TOF MS (DHB:LiCl:initiator 10:1:1).
:1:1).34 Reduced site saccharose-based initiator 2: M (+Li+) = 889 (5.8%), 981 (17.6%), 1.073 (27.6%), 1.165 (25.8%), 1.257 (14.5%), 1.348 (8.7%) g mol−1 by MALDI-TOF MS (DHB:LiCl:initiator 10:1:1).
Preparation of star-shaped teroligomers
A previous procedure34 for the preparation of poly(tert-butylmethacrylate) stars by ATRP was modified as follows: 0.20 g (0.13 mmol) 1, 1.76 g (10.4 mmol) GBLMA, 2.43 g (10.4 mmol) MAMA, 1.22 g (5.2 mmol) HAMA, 0.03 g (0.35 mmol) CuCl and 0.93 g (0.69 mmol) CuCl2 and 20 g anisole were added to a 50 ml round bottom flask sealed with a rubber septum, equipped with a stir bar, and purged with nitrogen for 30 min. The solution was heated in a 65 °C oil bath and 0.18 g (1.04 mmol) PMDETA were added at time t = 0. A characteristic color change to transparent green indicated the start of the reaction.Aliquots were taken throughout the reaction to follow the conversion of all three monomer with 1H-NMR simultaneously. Peak integration was performed on the vinyl peaks of GBLMA, MAMA, HAMA at a chemical shift of δ = 6.39 ppm, δ = 6.27 ppm and δ = 6.22 ppm, respectively, and calibrated to the integration of the anisole signal at δ = 3.87 ppm. The half-logarithmic relationship between conversion and polymerization time was used to determine the end time for 50% conversion (eqn (1)).35–37
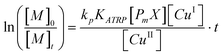 | (1) |
At the calculated end time, a final aliquot was removed to verify the final conversion and the reaction was quenched by rapid cooling to room temperature with liquid nitrogen. The reaction solution was passed through a silica column to remove the copper catalyst, concentrated under reduced pressure while being heated to 80 °C and precipitated in methanol. Following gravity filtration, the resulting powder was taken up in 1,4-dioxane and freeze dried to obtain the star-shaped (GBLMA-co-MAMA-co-HAMA) oligomer (S1d) as a white powder (2.75 g, 97%).
Similar procedures were taken out with reactant ratios and polymerization times modified to achieve samples with different defined arm lengths.
Gel permeation chromatography
GPC with a Wyatt DAWN HELEOS multiangle light scattering (GPC/MALS) detector equipped with a 632.8 nm He-Ne laser and Viscotek Model 250 viscosity detector (GPC/viscosity) were used to determine the absolute molecular weights. THF was used as eluent at a flow rate of 1.0 mL min−1: column set, 5 μm PSS SDV gel, 102, 103, 104, and 105 Å, 30 cm each. For GPC/viscosity, the refractive index increment, dn/dc, was measured on a PSS DnDC-2010/620 differential refractometer in THF at 25 °C.Lithographic experiments
For film preparation a 5 wt.-% solution of S1d and triphenylsulfonium perfluoro-1-butanoylsulfonate (19:1) in PGMEA was spin coated on a HMDS primed silicon wafer and prebaked at 125 °C for 150 s which corresponded to a 90 nm film thickness. The film was exposed with a 100 nm line/space (l s−1) pattern at doses from 10 to 410 μC cm−2 in 24 steps with a Zeiss 1530 FESEM equipped with a Raith Elphi Plus and an accelerating voltage of 20 kV. The film was annealed for post exposure bake (PEB) with a temperature gradient from 93 °C to 104 °C for 30 s and perpendicular to the temperature gradient developed in 0.26 N TMAH for four time periods of (15, 30, 60, 120) s. A Zeiss 1530 FESEM was used for imaging and the software SuMMIT™ was used for evaluation and determination of the line edge roughness (LER) and the line width roughness (LWR) of the l s−1 pattern.
Results and discussion
Analysis of initiator functionality
In order to achieve different functionalities from the same saccharose core active initiating groups were replaced with a non-reactive propylester resulting in reduced functionality. Scheme 1 shows the reaction pathway toward the nearly complete functionalized saccharose based initiator 1 and the initiator 2 with reduced functionality.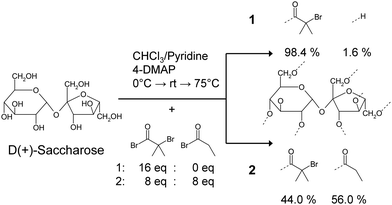 | ||
| Scheme 1 Reaction pathway toward saccharose based initiators 1 and 2. 1 is the maximum functionalized saccharose based initiator, while 2 has a reduced functionality realized by capping with the non-reactive propylester. | ||
Saccharose contains eight possible reaction sites for 2-bromoisobutyryl bromide and propionyl bromide. The synthetic route to initiator 1 yields the maximum functionalized saccharose based initiator while the synthesized initiator 2 is not a single initiator, but rather a mixture of initiators based on the same saccharose molecule, each of different functionality. The composition of the multifunctional initiators was analyzed by MALDI-TOF mass spectrometry (MS) to determine the numbers of initiating sites and the corresponding content in the mixture of the synthesized initiator 2 (Table 1).
| Mw (g mol−1)a | Initiating sitesb | Content (%)c |
|---|---|---|
| a Molecular weight of MALDI-TOF MS detected initiator + (Li+). b Number of brominated initiating sites determined by tabulation of molecular weight for possible combinations of sites reacted with 2-bromoisobutyryl bromide, sites reacted with propionyl bromide, and debrominated initiators yielded by the purification procedure. c Content of corresponding initiator derivative in the synthesized mixture of initiator 2. The content is calculated by the integrated strength of the MALDI-TOF MS peaks. | ||
| 889 | 1 | 5.8 |
| 981 | 2 | 17.6 |
| 1.073 | 3 | 27.6 |
| 1.165 | 4 | 25.8 |
| 1.257 | 5 | 14.5 |
| 1.348 | 6 | 8.7 |
The MALDI-TOF MS measurement shows a Gaussian content distribution with the main content in the mixture of initiator 2 between three (27.6%) and four (25.8%) initiating sites. As a result the initiator 2 has the overall average of 3.5 bromine based initiating sites which is given by the endcapping of the saccharose core with propionyl bromide and by the loss of bromides during purification procedure.
The efficiency of the reactive sites to initialize polymerization has to be taken into account, reducing the arm number further. A practical value of 66% is well-established by numerous reports on methacrylate synthesis with initiating sites of identical reactivity and was also taken into consideration for the initiator efficiency. Thus, a final number of arms of 5.3 for 1 is anticipated and in the same manner, the initiator efficiency of 2 is anticipated to be 2.4. With the initiator 1, the ethyl 2-bromoisobutyrate initiator and the initiator 2 we are allowed to synthesize polymers of a star architecture containing more than five arms, linear polymers as well as the corresponding missing links with more than two arms.
Monomer feed ratios
For this study, an average monomer composition of 50% GBLMA, 30% MAMA and 20% HAMA within the product was targeted due to its potential application as a photoresist material.30 Through repeated experimentation with a statistical linear copolymer, a feed of 41% GBLMA, 41% MAMA and 18% HAMA was found to achieve the target composition over a conversion range of Xp = 30–60% (Fig. 1). For this purpose, the composition was monitored throughout the polymerization by the decrease of the three vinyl signals, which corresponded to each monomer decaying in 1H-NMR. Except for the start of the reaction, the composition of the growing polymer remained close to the target composition. Conversion followed first order kinetics without any trace of side reactions like recombination or transfer. | ||
| Fig. 1 Evolution of monomer incorporation (GBLMA (◊), MAMA (□) and HAMA (○)) and also -ln(1-Xp) (★) versuspolymerization time of S1d. | ||
Synthesis of star-shaped teroligomers with modified arm lengths
Four star-shaped oligomers with varying target average arm lengths of 7, 10, 19 and 20 units and identical core 1, a star with modified core 2 and a linear model oligomer initiated from ethyl 2-bromoisobutyrate were synthesized (Scheme 2; Table 2). For all materials the average monomer composition is 50% GBLMA, 30% MAMA and 20% HAMA, which demonstrates the effectiveness of the proposed method. S1c shows the most distinct star character, a similar molecular weight and the same composition as a hyperbranched polymer published by Hadziioannou et al.26 Thus S1d was synthesized with a focus on the same molecular weight of S1c but in a larger scale for lithographic investigations. In addition, as shown above in Fig. 1 the desired composition is independent on the investigated conversions, this second batch was synthesized up to a higher conversion of 60% to save monomer. | ||
| Scheme 2 Synthesis of the linear model teroligomer L and the star-shaped teroligomers 1 and 2. The overall teroligomer composition of GBLMA/MAMA/HAMA (GcMcH) was 50/30/20. 1 is the maximum functionalized saccharose initiator, while 2 has a reduced functionality. | ||
| Code | Polymer a,b | [I]0(mmol L−1) | [M]0c:[I]0:[L]:[CuI]0:[CuII]0 | Time(min) | Mn, theo.d(kg mol−1) | Mn,MALS(kg mol−1) | PDIe | DPnf |
|---|---|---|---|---|---|---|---|---|
| a First subscript denotes degree of polymerization per arm. b Second subscript denotes arm number. c Monomer feed ratio: 41% GBLMA, 41% MAMA, 18% HAMA. d theoretical targeted molecular weight. e polydispersity index measured by MALS detection. f overall degree of polymerization deduced from MALS values. | ||||||||
| L | (GcMcH)41 | 27.0 | 55: 1.0: 1.0: 0.5:0.5 |
200 | 7.0 | 8.4 | 1.05 | 41 |
| S1a | ((GcMcH)7)5.3 | 31.4 | 70: 1.0: 8.0: 0.5: 7.5 | 120 | 8.4 | 9.1 | 1.05 | 38 |
| S1b | ((GcMcH)10)5.3 | 8.9 | 119: 1.0: 8.0: 1.66.4 | 105 | 12.0 | 12.5 | 1.05 | 55 |
| S1c | ((GcMcH)19)5.3 | 4.5 | 237: 1.0: 8.0: 4.0: 4.0 | 124 | 22.4 | 20.6 | 1.03 | 98 |
| S1d | ((GcMcH)20)5.3 | 6.5 | 200: 1.0: 8.0: 2.7: 5.3 | 150 | 22.4 | 23.9 | 1.05 | 104 |
| S2 | ((GcMcH)14)2,4 | 25.0 | 75: 1.0: 4.0: 0.8: 3.2 | 45 | 8.2 | 8.0 | 1.08 | 35 |
The ratio of monomer units to initiator plays a crucial role in synthesizing the desired arm length and the average monomer composition of the star. Reaction conditions were adjusted for each polymerization to result in monomer percentages of 50%, 30% and 20%, for GBLMA, MAMA and HAMA, respectively, when conversion approached the predetermined region of 40–60%. Since the reaction time decreases sharply when synthesizing shorter molecules, the ratio [CuI]0/[CuII]0 was decreased to slow down reaction times significantly. The addition of [CuII] deactivator allows targeting very short arms without losing control over the reaction. Analysis of the final aliquots yielded an overall composition of (46/32/22), (47/31/22), (50/31/19), (50/30/20), (49/30/21) and (49/31/20) (GBLMA%/MAMA%/HAMA%) for L, S1a, S1b, S1c, S1d and S2. This is very well within the expected range and underlines previously made assumptions.
The conversion index (eqn (1)) describes a logarithmic relationship between the conversion of monomer and polymerization time and therefore corresponds to a first order kinetic.35–37 By normalizing the conversion index with reaction quantities that are proportional to the remaining terms (eqn (2)), it can be shown that all four reactions follow the same kinetics (Fig. 2) independent of [I]0/[CuI]0 and [CuI]0/[CuII]0 ratios.
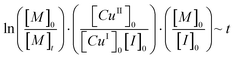 | (2) |
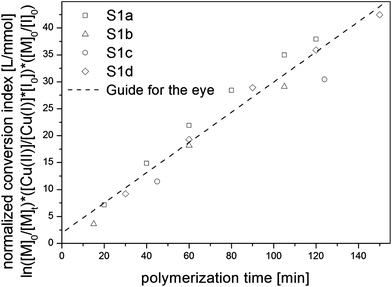 | ||
| Fig. 2 Plot of the normalized conversion index versuspolymerization time for S1a (□), S1b (△), S1c (○), S1d (◊). | ||
This information can then be used to predict how changing each reaction condition will impact the polymerization time required to achieve the desired conversion.
As already mentioned in the introduction star teroligomers consist of polymer chains covalently linked to one core resulting in reduced hydrodynamic radii compared to linear polymer coils, so standard GPC does not deliver reliable values for molecular weights without corresponding calibration. Thus GPC measurements were performed with a multi-angle light scattering (MALS) detector to obtain absolute molecular weights (Fig. 3).
 | ||
| Fig. 3 Normalized scattering intensity of GPC traces of L, S1a, S1b, S1c, S1d and S2 measured with THF as the eluent and a multi-angle light scattering detector. Slight recombination are visible by shoulders for the star-shaped teroligomers S1c (dash dot) and S1d (dash dot dot) due to the fact, that the scattering detector is disproportionally sensitive to the molecular size (intensity ∼ diameter6). | ||
As already indicated by the first order kinetics and observed for all star-shaped teroligormer narrow monomodal distributions were detected. All traces have a symmetrical shape and show no signs of tailing indicating a quantitative initiation and a controlled character. The shift to a lower elution volume corresponds to an increase in molecular weight and hence, an increasing arm length for the star teroligomers series S1a–S1d with an average of 5.3 arms. These traces are comparable in quality (shape and distribution) to their linear counterpart indicating equally good control. The scattering detector is disproportionally sensitive (Intensity ∼ diameter6)38 to changes in molecular size. Hence, recombination shoulders result in a disproportionally strong signal with MALS detection as can be seen for sample S1c (dash dot) and S1d (dash dot dot). As a result the GPC analysis shows negligible coupling up to 50% conversion. The reason for this rather unusual high conversion may be explained by the dilution of 5:1 excess anisole to monomer and especially by the steric hindrance of the bulky monomers suppressing termination reactions. According to evaluation of reaction kinetics and the fact that sterical hindrance of the bulky monomers suppresses termination reactions, the arm length has no influence on the extension of the coupling reactions. In summary, this series of star-shaped oligomers successfully demonstrates the strength of this synthetic route at preparing well-defined copolymers of diverse arm length but of the same monomer composition.
Using the normalized conversion index (eqn (2)), two different star-shaped teroligomers (S1a, S2) and the linear oligomer (L) were prepared with an overall target DPn of 40. This yielded three materials comprised of the same monomer compositions but different architectures. The resulting materials demonstrate the opportunity for fundamental studies. Series similar to this would allow detailed investigations of the impact of architecture to material properties in low molecular weight systems. The synthesis work demonstrates the reproducible reaction pathway and precise prediction capability for complex copolymers with different architecture.
The chemical nature and the resulting initiating efficiency, f, of initiators S1a (f = 5.3), S2 (f = 2.4), and L (f = 1) directly influences the slope of the normalized conversion index versus time, but show an almost perfect linear dependency (Fig. 4).
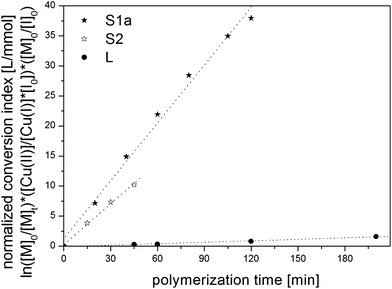 | ||
| Fig. 4 Plot of normalized conversion index versus time for S1a (★), S2 (☆), L (●). The different slopes originate from the different investigated initiators. | ||
In Fig. 4 the plot of the normalized conversion index and the represented linear connection shows impressively the independency of [I]0/[CuI]0 and [CuI]0/[CuII]0 ratios, but corresponding synthesis requires a various polymerization time obvious by the different slopes This allows the systematical investigation of S1a, S2 and L synthesized under different reaction conditions.
In order to achieve the highest level of precision, the individual conversion rate had to be determined for each initiator. Dilution and catalyst ratios can then be altered accordingly to achieve a polymerization of sufficient duration to take multiple 1H-NMR aliquots. As a consequence the investigated ATRP reaction follows the same kinetic and allows this systematic investigation of star polymers of different arm numbers and a linear model polymer.
Lithographic application
The literature known linear teroligomer synthesized from the monomers GBLMA, MAMA and HAMA is a high performance resist material and has recently been the focus of other research.39,40 In addition, Hadziioannou et al. published the first lithographic investigations on hyperbranched terpolymers of these monomers at the same copolymer composition.26 All of the synthesized resists with this specific monomer formulation had a targeted molecular weight of about 21 kg/mol. The patterning process of the hyperbranched polymer yielded a line edge roughness (LER) (3σ) of ∼6 nm and a line width roughness (LWR) (3σ) of ∼10 nm. Here, the well-defined star S1d in contrast to the non-controlled hyperbranched polymer is investigated for a proof of principle of utilizing the star architecture for lithographic patterning. The star teroligomer S1d is synthesized according to the above optimized formulation and has a comparable MW of 23 kg mol−1 to the published hyperbranched resist material. The optimization of the lithographic variables (post apply bake temperature, development time and exposure dose) for the new star resist were investigated with a combinatorial library: triphenylsulfonium perfluoro-1-butanoylsulfonate, a photoacid generator, was added to the star resist. This mixture was dissolved in propylene glycol monomethyl ether acetate and spin-cast. The post apply bake was done at 125 °C for 150 s on a hotplate to remove residual solvent and a film thickness of 90 nm was obtained. Electron beam lithography, a direct write technique, was used to generate an acid from the photo acid generator simulating a high resolution photolithography scanner. The exposed area consisted of a dose gradient (10–410 μC cm−2), exposing 100 nm line/space pattern on an area of (100 μm)2. A subsequent thermal treatment for 30 s (post exposure bake; PEB), performed with a temperature gradient from 95 °C to 104 °C, activated the acid catalyzed deprotection of the polymer component methyl adamantyl methacrylate releasing the polar carboxyl groups responsible for the development contrast. A development time gradient was achieved by dipping the resist film stepwise in a base developer (0.26 N tetramethylammonium hydroxide; TMAH) for (15, 30, 60, and 120) s to dissolve the exposed areas. By applying the three gradients in different directions to the same substrate a ternary combinatorial library schematically shown in Scheme 3 was prepared. Selected SEM images of line/space patterns and their corresponding LER and LWR values are depicted in Scheme 3 representing the 576 unique combinations of the aforementioned three variables. | ||
| Scheme 3 The upper figure shows a schematic illustration of the ternary combinatorial library consisting of the PEB temperature gradient, the development time step gradient, and the e-beam dose gradient. The table exhibits patterns from the four corners of the combinatorial library (I, II, IV, V) and the optimized pattern (III). The respective conditions (PEB temperature, development time, and dose) of these patterns and the calculated LER and LWR are listed. | ||
The combinatorial investigation demonstrates the strong interdependence of resist processing variables. The optimized patterns were observed at a PEB temperature of 98 °C, a development time of 60 s, and an e-beam dose of 214.2 μC cm−2 resulting in an excellent LER value of 3.1 nm and LWR of 5.9 nm. The patterns with a PEB temperature outside the observed optimum temperature exhibit visible increases of LER and LWR (image I and V). The investigated development time step gradient demonstrates the clear contrast of the star resist S1d. Here, the trend is observed that a short development time (image II and IV) results in clearer lines than those achieved after overdeveloping at 120s (image I and V). The electron beam dose exposure gradient shows the strong influence of this variable and its dependence on the other lithographic variables. Underexposure and overexposure was detected by sharp increases in LER and LWR values and by resist stripping. In conclusion, the combinatorial investigation results in an efficient optimization for the overall lithographic process variables in one library and demonstrates the high potential of the star resist as an architectural candidate for low roughness photoresists.
Conclusions
This work greatly expands both the precision of star-shaped oligomers synthesis and the capability to study the impact of arm number and arm length in the star-shaped oligomer regime for a wide range of materials and properties. Four star teroligomers with average arm lengths of 7, 10, 19, and 20 monomer units and a linear model oligomer were synthesized with an identical monomer composition by changing the ratios of [M]0/[I]0 and [CuI]0/[CuII]0. In addition, a technique for preparing multifunctional initiators with reduced functionality was introduced. High precision, star-shaped teroligomers initiated from the same core but with different numbers of arms were successfully synthesized. The achieved narrow and symmetrical molecular weight distributions with low polydispersity indices (PDIs < 1.1) were achieved even at conversion up to Xp = 50%. The potential of utilizing the well-defined star architecture for lithographic purposes was demonstrated by a combinatorial approach achieving a novel high resolution positive photoresist with LER of 3.1 nm and LWR of 5.8 nmAcknowledgements
This work was supported by the Semiconductor Research Corporation's Global Research Collaboration (GRC) Program [Tasks 1675.002 and 1677.001] and the German Research Foundation (Deutsche Forschungsgemeinschaft), Collaborative Research Center (Sonderforschungsbereich) 481, project A6. D.C.F. was supported by a GRC and Applied Materials fellowship. Research was performed in part at the University of Bayreuth, Macromolecular Chemistry I, the Bayreuth Institute of Macromolecular Research, and the Cornell Center for Materials Research, which is supported by NSF [Grant DMR 0520404] part of the NSF Materials Research Science and Engineering Center program. In addition we thank EUV technology for the roughness evaluation software SuMMIT™.References
- K. Matyjaszewski and N. V. Tsarevsky, Nat. Chem., 2009, 1, 276–288 CrossRef CAS.
- C. N. Likos and H. M. Harreis, Condens. Matter Phys., 2002, 29, 173–200 Search PubMed.
- C. N. Likos, Soft Matter, 2006, 2, 478–498 RSC.
- C. N. Likos, H. Löwen, M. Watzlawek, B. Abbas, O. Jucknischke, J. Allgaier and D. Richter, Phys. Rev. Lett., 1998, 80(20), 4450–4453 CrossRef CAS.
- M. Sawamoto, S. Kanaoka, T. Omura and T. Higashimura, Polym. Prepr., 1992, 33, 148–149 Search PubMed.
- R. J. Sutherland and R. B. Rhodes, Dispersant viscosity index improvers, 1994 Search PubMed , US5360564.
- H. J. Spinelli, Silicone containing acrylic star polymers, 1991, Search PubMed US5019628.
- R. J. Hunter, Foundations of Colloid Science, Vol. 1. Oxford University Press: New York, 1986 Search PubMed.
- G. Grest, L. J. Fetters and J. S. Huang, Adv. Chem. Phys., 1996, 94, 67–163 CAS.
- H.-P. Hsu, W. Nadler and P. Grassberger, Macromolecules, 2004, 37, 4658–4663 CrossRef CAS.
- C. von Ferber and Y. Holovatch, Condens. Matter Phys., 2002, 29, 117–136 Search PubMed.
- J. Dzubiella and A. Jusufi, Condens. Matter Phys., 2002, 30, 285–305 Search PubMed.
- J. Bohrisch, C. D. Eisenbach, W. Jaeger, H. Mori, A. H. E. Müller, M. Rehahn, C. Schaller, S. Traser and P. Wittmeyer, Adv. Polym. Sci., 2004, 165, 1–41 CAS.
- F. Ganazzoli, Condens. Matter Phys., 2002, 5, 37–71 Search PubMed.
- F. A. Plamper, H. Becker, M. Lanzendöfer, M. Patel, A. Wittemann, M. Ballauff and A. H. E. Müller, Macromol. Chem. Phys., 2005, 206, 1813–1825 CrossRef CAS.
- W. B. Liechty, D. R. Kryscio, B. V. Slaughter and N. A. Peppas, Annu. Rev. Chem. Biomol. Eng., 2010, 1, 149–173 Search PubMed.
- J. H. Adair, T. Li, T. Kido, K. Havey, J. Moon, J. Mecholsky, A. Morrone, D. R. Talham, M. H. Ludwig and L. Wang, Mater. Sci. Eng., R, 1998, 23, 1–242 Search PubMed.
- G. F. Matz, A. L. Melby, S.-R. T. Chen and N. F. Vozza, Low molecular weight water soluble polymer composition and method of use, 2001 Search PubMed , WO 2001006999.
- L. M. De Leon-Rodriguez, A. Lubag, D. G. Udugamasooriya, B. Proneth, R. A. Brekken, X. Sun, T. Kodadek and A. Dean Sherry, J. Am. Chem. Soc., 2010, 132, 12829–12831 CrossRef CAS.
- D. P. Sanders, Chem. Rev., 2010, 110, 321–360 CrossRef CAS.
- M. Yoshiiwa, H. Kageyama, F. Wakasa, M. Takai, K. Gamo and Y. Shirota, Appl. Phys. Lett., 1996, 69, 2605 CrossRef CAS.
- D. Yang, S. W. Chang and C. K. Ober, J. Mater. Chem., 2006, 16, 1693–1696 RSC.
- A. D. Silva and C. K. Ober, J. Mater. Chem., 2008, 18, 1903–1910 RSC.
- J. Dai, S. W. Chang, A. Hamad, D. Yang, N. Felix and C. K. Ober, Chem. Mater., 2006, 18, 3404–3411 CrossRef CAS.
- G. G. Barclay, M. A. King, Z. Mao, C. Szmanda, D. Benoit, E. Malmstrom, H. Ito and C. J. Hawker, Polym. Prepr., 1999, 40, 438–439 Search PubMed.
- C. L. Chochos, E. Ismailova, C. Brochon, N. Leclerc, R. Tiron, C. Sourd, P. Bandelier, J. Foucher, H. Ridaoui, A. Dirani, O. Soppera, D. Perret, C. Brault, C. A. Serra and G. Hadziioannou, Adv. Mater., 2009, 21, 1121–1125 CrossRef CAS.
- D. Baskaran, A. H. Mueller and K. Matyjaszewski, Controlled and Living Polymerizations: From Mechanisms to Applications, Wiley-VCH Verlag, Weinheim, Germany, 2010 Search PubMed.
- W. A. Braunecker and K. Matyjaszewski, Prog. Polym. Sci., 2007, 32, 93–146 CrossRef CAS.
- K. Matyjaszewski, ACS Symposium Series Vol. 1023, 2009.
- M. Zhang and A. H. E. Mueller, J. Polym. Sci., Part A: Polym. Chem., 2005, 33, 759–785 Search PubMed.
- D. M. Haddleton, R. Edmonds, A. M. Heming, E. J. Kelly and D. Kukulj, New J. Chem., 1999, 23, 477–479 RSC.
- G. Odian, Principles of Polymerization, John Wiley & Sons, Inc.: Hoboken, New Jersey, 2004 Search PubMed.
- D. Rein, P. Rempp and P. J. Lutz, Makromol. Chem., Macromol. Symp., 1993, 67, 237–249 Search PubMed.
- F. A. Plamper, H. Becker, M. Lanzendöfer, M. Patel, A. Wittemann, M. Ballauff and A. H. E. Müller, Macromol. Chem. Phys., 2005, 206, 1813–1825 CrossRef CAS.
- W. Tang and K. Matyjaszewski, Macromol. Theory Simul., 2008, 17, 359–375 CrossRef CAS.
- A. Goto and T. Fukuda, Prog. Polym. Sci., 2004, 29, 329–385 CrossRef CAS.
- K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101, 2921–2990 CrossRef CAS.
- H. Holthoff, S. U. Egelhaaf, M. Borkovec, P. Schurtenberger and H. Sticher, Langmuir, 1996, 12, 5541–5549 CrossRef CAS.
- H. Momose, A. Yasuda, A. Ueda, T. Iseki, K. Ute, T. Nishimura, R. Nakagawa and T. Kitayama, Proc. SPIE–Int. Soc. Opt. Eng., 2007, 6519, 65192F Search PubMed.
- K. Furukawa, S. Seki, T. Kozawa and S. Tagawa, Proc. SPIE–Int. Soc. Opt. Eng., 2008, 6923, 692334 Search PubMed.
Footnote |
| † Florian Wieberger and Drew C. Forman contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2012 |