O-Allylation of phenols with allylic acetates in aqueous media using a magnetically separable catalytic system†
Amit
Saha
,
John
Leazer
* and
Rajender S.
Varma
*
Sustainable Technology Division, National Risk Management Research Laboratory, U. S. Environmental Protection Agency, MS 443, Cincinnati, Ohio 45268, USA. E-mail: varma.rajender@.epa.gov; Fax: 513-569-7677; Tel: 513-487-2701
First published on 25th October 2011
Abstract
Allylic ethers were synthesized in water using magnetically recoverable heterogeneous Pd catalystvia O-allylation of phenols with allylic acetates under ambient conditions. The aqueous reaction medium, easy recovery of the catalyst using an external magnet, efficient recycling, and the high stability of the catalyst renders the protocol economic and sustainable.
Allyl ethers are important starting materials for a wide range of organic reactions, including the 1,3-hydrogen shift, [3,3]-sigmatropic rearrangement, and polymerization reactions,1 in addition to being a popular protecting group for alcohols.2 The traditional method for the synthesis of allyl ethers is the Williamson-type ether synthesis which involves the use of strongly basic metal alkoxide anions and highly active allyl halides or their equivalents.3 Comparatively, the addition of oxygen nucleophiles to η3-allylmetal complexes of various transition metals (Pd,4Ru,5Ir,6Ni7) provides an attractive mild alternative approach for their synthesis using much less reactive allyl alcohols, allyl esters or allyl carbonates. However, most of the existing methods for transition metal catalyzed O-allylationvia η3-allylmetal intermediates are accomplished using a non-recyclable homogeneous catalyst in various toxic organic solvents such as THF, DMF, benzene, toluene, and DCM under an inert atmosphere. The use of heterogeneous catalysts in organic transformations has become an interesting area of research in the field of green chemistry facilitating catalyst recyclability as exemplified in a recent report by Kobayashi et al.4a wherein a heterogeneous polymer incarcerated Pd catalyst is used for O-allylation of phenols in a THF medium. Magnetic nano-ferrite [Fe3O4] has been widely used by our group8 and others9 as the solid support for various catalysts because of their easy recovery using an external magnet. In continuation of the work with magnetically separable nano-ferrite [Fe3O4] as a heterogeneous catalyst support, Pd has been immobilized on the surface of dopamine-functionalized nanoparticles8a (Fig. 1), ([Fe3O4–dopamine–Pd]); their use as an easily recyclable catalyst to develop an economic and eco-friendly green synthesis of allyl ethers by O-allylation of phenols with allylic acetates in water under an open air atmosphere has been explored.
![[Fe3O4–dopamine–Pd] catalyst.](/image/article/2012/GC/c1gc16174a/c1gc16174a-f1.gif)
Phenols undergo unprecedented allylic substitution reactions with various allylic acetates in air under refluxing aqueous media conditions for 3–10 h whilst in the presence of the [Fe3O4–dopamine–Pd] catalyst; a mild base, sodium bicarbonate, is adequate to produce the allyl ethers (Scheme 1) in good yields.
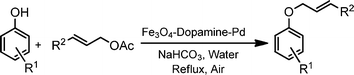 | ||
| Scheme 1 O-Allylation of phenols with allylic acetates. | ||
The catalyst was prepared following the optimized protocol established earlier.8a–c The aqueous suspension of nano-ferrite was sonicated for 2 h with dopamine, which acts as a pseudo-ligand and as a robust anchor preventing the leaching of immobilized Pd. Dopamine functionalized nano-ferrite was then treated with PdCl2 in basic medium to obtain the Pd(II) catalyst supported on amine functionalized magnetic Fe3O4 nanoparticles. The spherical morphology and the size of the nano catalyst (13–38 nm) were confirmed by transmission electron microscopy (TEM) (Fig. 2). The weight percentage of palladium was found to be 10.23% by inductively coupled plasma–atomic emission spectroscopy (ICP–AES) analysis.
![TEM image of [Fe3O4–dopamine–Pd] catalyst.](/image/article/2012/GC/c1gc16174a/c1gc16174a-f2.gif)
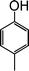
To optimize the reaction conditions, a series of experiments were conducted with a representative reaction of cinnamyl acetate and p-cresol, with variation of reaction parameters, such as base, solvent, reaction temperature etc. (Table 1). It was found that a combination of [Fe3O4–dopamine–Pd], 50 mg (4.8 mol%), and NaHCO3 (2 equiv.) in water is ideal for a fast and efficient reaction (Table 1, entry 13). The reaction was observed to be incomplete in the presence of a lower amount of the Pd catalyst (30 mg), even after prolonging the reaction time. Further, no improvement was observed by increasing the amount of Pd catalyst in terms of reaction time and yield of the product. The reaction does not proceed in either the absence of base NaHCO3 (Table 1, entry 1) or the catalyst [Fe3O4–dopamine–Pd] (Table 1, entry 2). Also, no desired allyl ether was obtained in an identical reaction with [Fe3O4] nanoparticles (without Pd) (Table 1, entry 3). Water was found to be the best solvent for this reaction to obtain improved yields and fast reaction times compared to other common organic solvents such as DMF, THF, CH3CN, toluene, PEG (Table 1, entries 4–7, 9).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Base | Solvent | Temp (°C) | Time (h) | Yield (%)b |
| a A mixture of p-cresol (1 mmol), cinnamyl acetate (1 mmol), base (2 mmol) and Pd catalyst (50 mg, 4.8 mol%) was heated under air in the specified solvent. b Yields refer to those of column purified isolated products. c The reaction was performed without the catalyst [Fe3O4–dopamine–Pd]. d The reaction was performed with Fe3O4 naoparticles (without Pd). | |||||
| 1 | — | Water | 100 | 15 | 0 |
| 2c | NaHCO3 (without catalyst) | Water | 100 | 15 | 0 |
| 3d | NaHCO3 (with Fe3O4) | Water | 100 | 15 | 0 |
| 4 | K2CO3 | DMF | 120 | 12 | 77 |
| 5 | K2CO3 | THF | 66 | 10 | 53 |
| 6 | K2CO3 | CH3CN | 82 | 10 | 55 |
| 7 | K2CO3 | Toulene | 100 | 12 | 9 |
| 8 | K2CO3 | Water | 100 | 10 | 80 |
| 9 | K2CO3 | PEG | 100 | 12 | 63 |
| 10 | Cs2CO3 | Water | 100 | 10 | 65 |
| 11 | Na2CO3 | Water | 100 | 10 | 80 |
| 12 | NaHCO3 | Water | 100 | 10 | 83 |
| 13 | NaHCO 3 | Water | 100 | 5 | 85 |
| 14 | NaOAc | Water | 100 | 10 | 18 |
| 15 | NaH | DMF | 120 | 12 | Trace |
| 16 | K3PO4 | Water | 100 | 10 | 66 |
In a typical representative experimental procedure, a mixture of allyl acetate, phenol, Pd catalyst and NaHCO3 in water was heated to reflux in an open atmosphere for a sufficient time to complete the reaction (as determined by TLC). After completion of the reaction, the catalyst was separated from the reaction mixture using an external magnet. The product was extracted by ethyl acetate and was purified by column chromatography. The catalyst was washed with acetone, dried under vacuum, and recycled for 5 consecutive reactions without any significant loss in efficiency (Table 2).
|
|
|
|---|---|
| No. of cycles | Yield (%) |
| 1 | 85 |
| 2 | 84 |
| 3 | 85 |
| 4 | 83 |
| 5 | 82 |
| 6 | 82 |
Metal leaching was studied by ICP–AES analysis of the catalyst before and after the reaction cycles. The Pd concentration in the heterogeneous catalyst was found to be the same before and after the reaction. The TEM image of the catalyst taken after the fifth cycle of the reaction does not show any significant changes in the morphology and the size of the catalyst nanoparticles (15–37 nm) (Fig. 3), which indicates retention of the catalytic activity after recycling. No Pd metal was detected in the reaction solvent (water) after completion of the reaction. It confirms the fact that dopamine-functionalization provides enough amine-binding sites on the surfaces of the Fe3O4 nanoparticles, which serve as pseudo-ligands by coordinating with Pd and thus help to minimize deterioration and metal leaching, whilst aiding in efficient catalyst recycling.

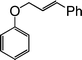
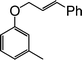
A series of substituted cinnamyl acetates underwent coupling with a variety of substituted phenols by this procedure to produce the corresponding allyl aryl ethers (Table 3). In general, the reactions are very efficient and clean. The ensuing products are obtained in high yields and good purity. Several functional groups such as OMe, NO2, Cl, and Br are compatible with this reaction, which performs consistently with o-, m-, and p-substituted phenols (Table 3, entries 2–4). A branched allylic acetate (Table 3, entry 13) was used with equal efficiency to provide a linear allyl aryl ether, supporting a mechanism that entails the intermediacy of an η3-allyl Pd complex as the key intermediate in the reaction (Scheme 2).
| Entry | Phenols | Allylic acetate | Time (h) | Products | Yields (%)b |
|---|---|---|---|---|---|
| a A mixture of phenol (1 mmol), allylic acetates (1 mmol), sodium bicarbonate (2 mmol) and Pd catalyst (50 mg, 4.8 mol%) in water (2.5 mL) was heated to reflux in air. b Yields refer to those of column purified isolated products. c The reaction mixture in DMF (2 mL) was heated at 90 °C. | |||||
| 1 |
|

|
6 |
|
80 |
| 2 |

|

|
5 |

|
85 |
| 3 |

|

|
5 |
|
87 |
| 4 |

|
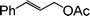
|
5 |

|
83 |
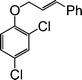
| 5c |
|

|
10 |
|
90 |
| 6 |
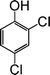
|

|
9 |
|
79 |
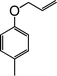
| 7 |
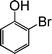
|
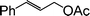
|
10 |
|
85 |
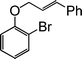
| 8 |
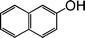
|
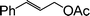
|
3 |
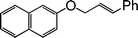
|
75 |
| 9 |
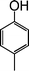
|
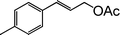
|
8 |
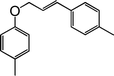
|
87 |
| 10 |
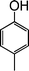
|
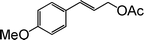
|
8 |
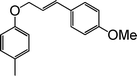
|
75 |
| 11 |
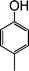
|

|
8 |

|
73 |
| 12 |
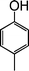
|
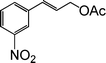
|
8 |

|
76 |
| 13 |

|

|
8 |
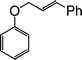
|
81 |
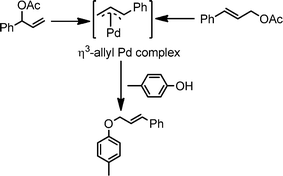 | ||
| Scheme 2 Formation of η3-allyl Pd complex as reaction intermediate. | ||
Conclusions
In conclusion, a simple and efficient green procedure for the general O-allylation of phenols with allyl acetates has been developed using a magnetically separable and easily recyclable heterogeneous Pd catalyst in the presence of a mild base, NaHCO3, in water under an open atmosphere. This precludes the requirement of using an inert atmosphere and organic solvents. Easy magnetic separation of the catalyst eliminates the requirement of catalyst filtration after completion of the reaction, which is an additional sustainable aspect of this reaction. To the best of our knowledge, this is the first example of an O-allylation reaction using a magnetic heterogeneous Pd catalyst in water.Experimental
O-Allylation of phenols by allylic acetates using [Fe3O4–dopamine–Pd] catalyst
A mixture of phenol (1 mmol), allylic acetate (1 mmol), NaHCO3 (2 mmol) and Pd catalyst (50 mg, 4.8 mol%) was added to 2.5 mL of water. The aqueous reaction mixture was heated to reflux for the required time as indicated in Table 3. The progress of the reaction was monitored by TLC. Standard work-up with ethyl acetate followed by simple column chromatography provided the pure product. All products listed in Table 3 were known in the literature (except entries 6 and 11) and were identified by comparison of their FT-IR, 1H, and 13C NMR with literature data. The products of entries 6 and 11, Table 3, were characterized by FT-IR, 1H and 13C NMR spectroscopy and elemental analysis.Acknowledgements
Amit Saha was supported by the Postgraduate Research Program at the National Risk Management Research Laboratory administered by the Oak Ridge Institute for Science and Education through an inter-agency agreement between the U. S. Department of Energy and the U. S. Environmental Protection Agency.References
- (a) J. Tsuji, in Handbook of Organopalladium Chemistry for Organic Synthesis Vol. 5 (Ed.: E. Negishi), Wiley, New York, 2002, pp. 1669–1687 Search PubMed; (b) B. M. Trost and D. L. VanVranken, Chem. Rev., 1996, 96, 395–422 CrossRef CAS; (c) S. A. Godleski, in Comprehensive Organic Synthesis Vol. 4 (ed.: B. M. Trost, I. Fleming), Pergamon, Oxford, 1991, pp. 585–661 Search PubMed.
- S. Tanaka, H. Saburi, Y. Ishibashi and M. Kitamura, Org. Lett., 2004, 6, 1873–1875 CrossRef CAS and references therein.
- A. W. Williamson, J. Chem. Soc., 1852, 4, 229 Search PubMed.
- (a) R. Akiyama and S. Kobayashi, J. Am. Chem. Soc., 2003, 125, 3412–3413 CrossRef CAS; (b) T. Satoh, M. Ikeda, M. Miura and M. Nomura, J. Org. Chem., 1997, 62, 4877–4879 CrossRef CAS; (c) C. Goux, M. Massacret, P. Lhoste and D. Sinou, Organometallics, 1995, 14, 4585–4593 CrossRef CAS; (d) Y. Kayaki, T. Koda and T. Ikariya, J. Org. Chem., 2004, 69, 2595–2597 CrossRef CAS; (e) H. Kim and C. Lee, Org. Lett., 2002, 4, 4369–4371 CrossRef CAS; (f) F. L. Lam, T. T.-L. Au-Yeung, F. Y. Kwong, Z. Zhou, K. Y. Wong and A. S. C. Chan, Angew. Chem., Int. Ed., 2008, 47, 1280–1283 CrossRef CAS; (g) J. Muzart, J. Organomet. Chem., 1987, 326, C23–C28 CrossRef CAS; (h) C. Goux, P. Lhoste and D. Sinou, Synlett, 1992, 725–727 CrossRef CAS; (i) R. Lakhmiri, P. Lhoste and D. Sinou, Tetrahedron Lett., 1989, 30, 4669–4672 CrossRef CAS.
- (a) H. Saburi, S. Tanaka and M. Kitamura, Angew. Chem., Int. Ed., 2005, 44, 1730–1732 CrossRef CAS; (b) Z. Sahli, N. Derrien, S. Pascal, B. Demerseman, T. Roisnel, F. Barriere, M. Achard and C. Bruneau, Dalton Trans., 2011, 40, 5625–5630 RSC; (c) J. A. v. Rijn, E. v. Stapele, E. Bouwman and E. Drent, J. Catal., 2010, 272, 220–226 CrossRef; (d) R. C. v. d. Drift, M. Vailati, E. Bouwman and E. Drent, J. Mol. Catal. A: Chem., 2000, 159, 163–177 CrossRef; (e) J. A. v. Rijn, M. A. Siegler, A. L. Spek, E. Bouwman and E. Drent, Organometallics, 2009, 28, 7006–7014 CrossRef; (f) M. Austeri, D. Linder and J. Lacour, Adv. Synth. Catal., 2010, 352, 3339–3347 CrossRef CAS.
- H. Nakagawa, T. Hirabayashi, S. Sakaguchi and Y. Ishii, J. Org. Chem., 2004, 69, 3474–3477 CrossRef CAS.
- Y. Yatsumonji, Y. Ishida, A. Tsubouchi and T. Takeda, Org. Lett., 2007, 9, 4603–4606 CrossRef CAS.
- (a) V. Polshettiwar and R. S. Varma, Org. Biomol. Chem., 2009, 7, 37–40 RSC; (b) V. Polshettiwar and R. S. Varma, Chem.–Eur. J., 2009, 15, 1582–1586 CrossRef CAS; (c) V. Polshettiwar, B. Baruwati and R. S. Varma, Green Chem., 2009, 11, 127–131 RSC; (d) V. Polshettiwar, B. Baruwati and R. S. Varma, Chem. Commun., 2009, 1837–1839 RSC; (e) R. Luque, B. Baruwati and R. S. Varma, Green Chem., 2010, 12, 1540–1543 RSC.
- (a) C. Dalaigh, S. A. Corr, Y. Gun'ko and S. J. Connon, Angew. Chem., Int. Ed., 2007, 46, 4329–4332 CrossRef CAS; (b) R. L. Oliveira, P. K. Kiyohara and L. M. Rossi, Green Chem., 2010, 12, 144–149 RSC; (c) Z. Yinghuai, L. Kuijin, N. Huimon, L. Chuanzhao, L. P. Stubbs, C. F. Siong, T. Muihua and S. C. Peng, Adv. Synth. Catal., 2009, 351, 2650–2656 CrossRef; (d) C. Che, W. Li, S. Lin, J. Chen, J. Zheng, J.-C. Wu, Q. Zheng, G. Zhang, Z. Yang and B. Jiang, Chem. Commun., 2009, 5990–5992 RSC; (e) K.-I. Fujita, S. Umeki, M. Yamazaki, T. Ainoya, T. Tsuchimoto and H. Yasuda, Tetrahedron Lett., 2011, 52, 3137–3140 CrossRef CAS; (f) T. Zeng, W.-W. Chen, C. M. Cirtiu, A. Moores, G. Song and C.-J. Li, Green Chem., 2010, 12, 570–573 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures; 1H and 13C NMR spectra of all products. See DOI: 10.1039/c1gc16174a |
| This journal is © The Royal Society of Chemistry 2012 |