Torrefaction of Loblolly pine
Haoxi
Ben
and
Arthur J.
Ragauskas
*
School of Chemistry and Biochemistry, Institute of Paper Science and Technology, Georgia Institute of Technology, Atlanta, Georgia 30332, USA. E-mail: Arthur.ragauskas@chemistry.gatech.edu; Fax: (+1) 404-894-4778; Tel: (+1) 404-894-9701
First published on 31st October 2011
Abstract
The torrefaction of Loblolly pine (Pinus taeda) was examined at 250 and 300 °C, to determine the effects of treatment temperatures on the chemical structure of the torrefied Loblolly pine. Solid-state cross-polarization/magic angle spinning (CP/MAS) 13C nuclear magnetic resonance (NMR) spectroscopy was used to characterize the torrefied and native Loblolly pine. The NMR results indicate that aryl-ether bonds in lignin were cleaved during the torrefaction. The methyl carbons in hemicellulose acetyl groups were no longer present after the torrefaction at 250 °C for 4 h, which is consistent with HPLC carbohydrate analysis of the torrefied wood which indicated that the hemicellulose fraction of pine was completely absent, whereas the cellulose and lignin remained largely intact. Under these conditions the torrefied wood has a relatively high energy yield of 81.29% and a HHV of 24.06 MJ kg−1. After torrefaction at 300 °C for 4 h, the cellulose and hemicellulose in the wood were completely eliminated, the residue contains enriched amounts of carbonyl groups, aromatic carbons and methoxyl groups, which represent complex condensed aromatics, these aromatics units were linked with aliphatic C–O and C–C bonds and the product has a very high HHV of 32.34 MJ kg−1.
Worldwide energy consumption is predicted to increase by ∼50% by 2025.1 The carbon dioxide emissions from the consumption of fossil fuels have grown at an average rate of ∼2% per year, and the rate continues to increase. Coal is the most carbon-intensive of the fossil fuels and its global share of carbon dioxide emissions, are projected to be almost 46% by 2035.2 Growing concerns about the effects of carbon dioxide emissions from fossil fuels call for sustainable energy sources, such as biomass, because of its carbon neutrality, relative abundance and non-food competition.3 Several challenging properties of biomass including low heating value, low energy density, high moisture content, hygroscopic nature, and soot formation during combustion limit its usage as a resource for the bio-energy.4 The charcoal-making process is one of the traditional methods to upgrade the biomass. However, only about 20–55% of the energy in the original raw material is retained in charcoal.5 In contrast, a thermal treatment process in an inert atmosphere, known as torrefaction, can address some of the above limitations and provide a relatively higher energy yield.6
Many researchers have examined the torrefaction of biomass. For example, Pentananunt et al.5 found that torrefied wood has significantly less soot generation during combustion and has a relatively faster rate of combustion than the wood. The weight and energy yields of torrefaction of wood at 250–270 °C for 2–3 h are 66.7–83.3% and 76.5–89.6%, respectively. Prins et al.7,8 examined the weight loss kinetics and analyzed the products of torrefaction of several bioresources including beech, willow, larch and wheat straw. They proposed torrefaction of wood is a two step reaction, a fast initial step leads to the decomposition of hemicelluloses, and the slower subsequent reaction represents cellulose decomposition. Yan et al.9 examined wet torrefaction (hot compressed water, 200–260 °C, ∼20 min) and the dry torrefaction (nitrogen, 250–300 °C ∼80 min) of Loblolly pine. For the dry torrefaction, the mass yields of solid product were 61–84%, and energy densities increased by 7–21%. They also found that the decomposition of hemicellulose occurs more readily during wet torrefaction than dry torrefaction. Chen et al.10 torrefied four kinds of biomass, including bamboo, willow, coconut shell and wood (i.e. Ficus benjamina L.). Similarly, with the results proposed by Prins et al.,7,8 they also found that there are two different torrefaction processes, regardless of the biomass resource, involving a light torrefaction, which involved significant decomposition of hemicellulose, at 240 °C. Subsequent severe torrefaction of cellulose occurs at 275 °C. Pimchuai et al.4 examined the torrefaction of rice husks and four other agriculture residues (i.e., sawdust, peanut husks, bagasse, and water hyacinth) at 250–300 °C for 1–2 h. They found that compared to the raw biomass, higher heating values (HHV) were obtained for the torrefied biomass by 7–40%. The hydrophobic properties of torrefied biomass were markedly improved with respect to the starting material which means the torrefied biomass absorbs much less moisture during storage. Chen et al.11 torrefied some basic biomass constituents, including hemicellulose, cellulose, lignin, xylan, dextran, xylose and glucose at 230, 260 and 290 °C by thermogravimetry. They indicated that torrefaction at 230 °C has a relatively slight impact on decomposing basic biomass components, 260 °C caused a certain amount of hemicellulose decomposed, and at 290 °C a large amount of decomposition of hemicellulose and cellulose occurred. Brosse et al.12 thermally treated beech heartwood (Fagus sylvatica) at 230 °C for 7 h, they indicated that thermally treatment of wood could degrade the hemicelluloses and cleave the β-aryl-ether linkages in lignin, and condensed aromatic units will be formed by condensation reactions of lignin and cellulose decomposition products. Pastorova et al.13 also reported a similar highly condensed aromatic polymer. They examined the structure of char produced by thermal treatment of cellulose at 190–390 °C and concluded that disproportionation occurred above ∼310 °C and lead to a highly condensed aromatic polymer.
In summary, although process parameters of biomass torrefaction have been studied the chemistry of torrefaction on wood and other bioresources has received little attention. The goal of this work is examine the efforts of torrefaction at different temperatures and times on the chemical structures of torrefied wood derived from Loblolly pine. This was accomplished by using solid-state cross-polarization/magic angle spinning (CP/MAS) 13C nuclear magnetic resonance (NMR) spectroscopy and carbohydrate analysis.
Experimental
Wood chips used in this study were acquired from a 15-year old Loblolly pine tree from the southeastern US. Detailed information about this material has been reported by David et al.14 The wood chips were refined with a Wiley mill through a 0.13 cm screen. The refined wood had an 11.11 wt% moisture content and samples were stored at ∼0 °C prior to use. Torrefaction experiments were conducted in a quartz torrefaction tube heated with a split-tube furnace. Typically, the torrefaction sample (∼5.00 g) was placed in a quartz sample boat that was then positioned in the center of torrefaction tube. The torrefaction tube was flushed with nitrogen gas and the flow rate was adjusted to a value of 500 mL min−1 and then inserted in the pre-heated furnace. Upon completion of torrefaction, the reaction tube was removed from the furnace and cooled to room temperature under constant N2 flow. The torrefied wood was collected for subsequent chemical analysis and the yield was determined gravimetrically.The higher heating values of torrefied wood samples were measured in a Parr 1261 isoperibol bomb calorimeter according to literature methods.15 The HHVs reported in this study are based on the average of duplicated tests. The error is 0.6%.
Monosaccharide and Klason lignin content of the native wood and torrefied wood samples were analyzed by Dionex chromatography, high performance anion exchange chromatography with pulsed amperometric detection (HPAEC–PAD) for monosaccharide analysis on the basis of reported methods.15 The acid insoluble residue was reported as the Klason lignin and char content of the native wood and torrefied wood samples.
Solid-state CP/MAS 13C NMR was carried out using a Bruker DSX 300 NMR spectrometer operating at 75.48 MHz. The experiments were performed at ambient temperature using a Bruker 7 mm MAS probe. The samples were ground and packed into 7 mm ZrO rotors, which were spun at 4 kHz.14
Results and discussion
The mass yields, HHVs, energy densification ratios and energy yields of the torrefied wood samples produced from Loblolly pine wood at 250 and 300 °C are summarized in Table 1. The results indicate that the torrefied wood has a higher HHV than the original wood. Compared to the HHV (20.16 MJ kg−1) of dried wood (dried at 75 °C, 48 h), the HHV of torrefied wood increased by 60% after torrefaction at 300 °C for 4 h, which (32.34 MJ kg−1) is higher than many commercial coals, such as, anthracite coal (31.84 MJ kg−1) and Pittsburgh seam coal (31.75 MJ kg−1), and much higher than Converse School—Sub C coal (21.67 MJ kg−1), German Braunkohole lignite (25.10 MJ kg−1), and Northumerland No.81/2 Sem. Anth. Coal (24.73 MJ kg−1).16 For the torrefied wood samples produced at 250 °C, with an increase residence time from 0.25 to 8 h, the mass yield and energy yield decreased linearly from 94.97% to 64.36% and 99.79% to 79.12% respectively. In contrast, the HHV increased from 21.22 to 24.78 MJ kg−1. The mass yields of torrefied wood samples decreased significantly from 250 to 300 °C as there was less than 50 wt% of biomass after torrefaction at 300 °C, which is similar with the literature reports.4,11,17 Those dramatic differences between the torrefaction at 250 and 300 °C could be explained by one of the major wood components, cellulose, being decomposed near 300 °C.4,7,8,11,17,18 In contrast with the energy yields of torrefied wood samples produced at 250 °C, the energy yields increased with an increased torrefaction time at 300 °C, which indicate that prolonged treatment of wood at 300 °C will produce a material with a much higher energy density. These results indicate that the torrefaction of Loblolly pine wood at 250 °C for 4 h are the optimal conditions, which will produce a torrefied wood sample with an HHV of 24.06 MJ kg−1 and a relatively high energy yield of 81.29%, prolonged treatment only slightly increased the HHV of the torrefied wood.| T/°C | Time (h) | Mass yielda (%) | HHV (MJ kg−1) | Energy densification ratiob | Energy yieldc (%) |
|---|---|---|---|---|---|
| a Mass yield = mass of dried torrefied wood/mass of dried wood ×100%. b Energy densification ratio = HHV of dried torrefied wood/HHV of dried wood. c Energy yield = mass yield × energy densification ratio. d The original Loblolly pine wood sample was dried at 75 °C for 48 h before the analysis of higher heating value. | |||||
| Orignal pined | — | — | 20.16 | — | — |
| 250 | 0.25 | 94.79 | 21.22 | 1.05 | 99.79 |
| 0.50 | 86.19 | 21.87 | 1.08 | 93.48 | |
| 1.00 | 80.77 | 22.18 | 1.10 | 88.88 | |
| 2.00 | 75.46 | 22.61 | 1.12 | 84.62 | |
| 4.00 | 68.11 | 24.06 | 1.19 | 81.29 | |
| 6.00 | 66.19 | 24.40 | 1.21 | 80.11 | |
| 8.00 | 64.36 | 24.78 | 1.23 | 79.12 | |
| 300 | 0.50 | 45.74 | 23.10 | 1.15 | 52.41 |
| 1.00 | 40.36 | — | — | — | |
| 2.00 | 37.61 | — | — | — | |
| 4.00 | 36.65 | 32.34 | 1.60 | 58.79 |
The carbohydrate profiles of each torrefied wood sample are shown in Table 2. For the torrefied wood samples produced at 250 °C the content of glucose in those samples varied less than 20%. In contrast, the contents of arabinose, galactose, xylose and mannose linearly decreased with an increase in torrefaction time. The contents of arabinose and galactose approached zero and the contents of xylose and mannose decreased by almost 90% after a 4 h torrefaction at 250 °C. Therefore at the optimal conditions, almost all the hemicelluloses will be decomposed, whereas the cellulose remained largely intact. In contrast, the contents of glucose in the torrefied wood samples produced at 300 °C dramatically decreased. Employing a 4 h torrefaction treatment, the contents of all the monosaccharides were not detected (see Table 2) which indicates that all cellulose and hemicellulose in the wood were decomposed.
| T/°C | Time (h) | Glucose | Arabinose | Galactose | Xylose | Mannose | Klason lignin or char | Sum |
|---|---|---|---|---|---|---|---|---|
| a The results were shown as the weight percentage of sample. b The content of the acid soluble lignin is less than 0.7% in the original wood, which is not included in this table. | ||||||||
| Original wood | — | 47.89 | 1.13 | 2.35 | 6.20 | 10.60 | 28.50b | 96.67 |
| 250 | 0.25 | 46.27 | 0.66 | 1.94 | 4.64 | 9.19 | 32.20 | 94.90 |
| 0.50 | 48.42 | 0.28 | 1.18 | 2.89 | 6.55 | 37.25 | 96.57 | |
| 1.00 | 49.37 | 0.16 | 0.75 | 2.10 | 4.64 | 41.62 | 98.64 | |
| 2.00 | 47.71 | 0.00 | 0.38 | 1.26 | 2.83 | 44.53 | 96.71 | |
| 4.00 | 44.24 | 0.00 | 0.10 | 0.50 | 1.15 | 52.33 | 98.32 | |
| 6.00 | 43.88 | 0.00 | 0.00 | 0.46 | 1.00 | 55.40 | 100.74 | |
| 8.00 | 38.31 | 0.00 | 0.00 | 0.22 | 0.49 | 57.80 | 96.82 | |
| 300 | 0.50 | 22.34 | 0.00 | 0.14 | 0.49 | 1.22 | 73.39 | 97.58 |
| 4.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 99.43 | 99.43 |
To fully characterize the chemical structures of the torrefied wood and to understand the chemical changes occurring, CP/MAS 13C NMR was used to analyze the starting and torrefied wood samples as shown in Fig. 1 and 2. The NMR chemical shifts assignments of pine based on literature values are shown in Table 3.14,19–22
| Functional groups | Chemical shift (ppm) |
|---|---|
| a Signals for hemicellulose overlap with those peaks. b Signals for lignin overlap with those peaks. | |
| Carbonyl | 220–187 |
| Carboxyl | 173 |
| Aromatic C-3 or C-4 in guaiacyl lignin (etherified) | 153 |
| Aromatic C-3 or C-4 bond in guaiacyl lignin with free phenolic groups | 148 |
| Aromatic C–C bond in lignin | 131–137 |
| Aromatic C–H bond in lignin (G6 of lignin) | 120 |
| Aromatic C–H bond in lignin (G5 of lignin) | 115 |
| Aromatic C–H bond in lignin (G2 of lignin) | 112 |
| C-1 of cellulosea | 105 |
| Crystalline C-4 of cellulosea,b | 89 |
| Amorphous C-4 of cellulosea,b | 84 |
| C-2,C-3,C-5 of cellulosea,b | 75–72 |
| C-6 of cellulosea,b | 65–62 |
| Methoxyl in lignin | 56 |
| Methylene carbons CH2 in aliphatic chain (fatty acids and tannins)b | 32 |
| Methyl carbons CH3 in hemicellulose acetyl groupsb | 23 |
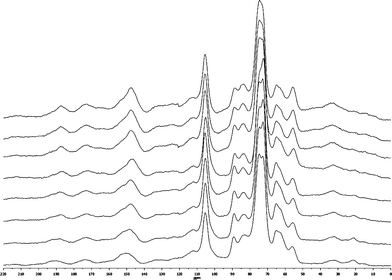 | ||
| Fig. 1 CP/MAS 13C NMR spectra (from bottom to top) of original Loblolly pine wood, torrefied wood samples produced by torrefaction of Loblolly pine wood at 250 °C for 0.25, 0.50, 1.00, 2.00, 4.00, 6.00 and 8.00 h. | ||
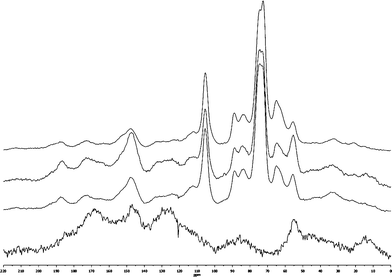 | ||
| Fig. 2 CP/MAS 13C NMR spectra (from bottom to top) of torrefied Loblolly pine wood samples produced by torrefaction of Loblolly pine wood at 300 °C for 4.00 h, 250 °C for 4.00 h, 300 °C for 0.50 h and 250 °C for 0.50 h. | ||
The relative signal intensities of the carbonyl and carboxyl groups slightly increased after torrefaction at 250 °C, which indicates the formation of those functional groups during the torrefaction.12 The peak centered at ∼148–153 ppm represents aromatic C–O bonds (ether bonds at ∼153 ppm and free phenolic hydroxyl groups at ∼148 ppm) in lignin, and during torrefaction, this peak shift from ∼152 to ∼148 ppm, and the intensity of the peak increases with an increased thermal treatment time, which indicates that there are more free phenolic hydroxyl groups after the torrefaction of wood. On the basis of the proposed cleavage pathway of ether bonds (see Fig. 3) in lignin during the thermal treatment, the cleavage of ether bonds will produce phenolic hydroxyl groups.12,23,24 Considering the results of CP/MAS 13C NMR, the increasing intensity of the peak at ∼148 ppm indicates that aryl ether bonds in lignin, such as β-O-aryl the linkages, were cleaved during the torrefaction. The signal intensities of aromatic C–C bonds and aromatic C–H bonds in lignin also increased after the torrefaction, which could be attributed to several factors, including the cleavage of lignin ether bonds during the thermal treatment which could re-condense to form aromatic C–C bonds.23,24 The thermal treatment of wood could also converted some carbohydrates to the aromatic C–C and C–H bonds, the proposed possible pathway of this conversion is shown in Fig. 3.12,25 The intensity of crystalline C-4 of cellulose at ∼89 ppm decreased upon thermal treatment which was in contrast to the intensity of the signal assigned to amorphous C-4 cellulose at ∼84 ppm. The latter signal increased with an increasing thermal treatment time from 4–8 h at 250 °C. However, due to unknown effects of the overlapping signals from hemicellulose and lignin side chains this observation can be attributed, only in part, as indicative of a reduction in the crystallinity of cellulose after the thermal treatment of wood. Ates et al.26 used FT-IR to determine the relative crystallinity indexes of the heat-treated (at 130, 180 and 230 °C) Calabrian pine wood and reported a similar result.
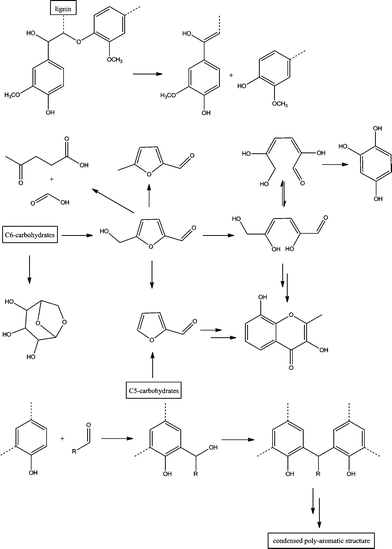 | ||
| Fig. 3 Hypothesized pathways of decomposition of major wood components and the formation of new functional groups and condensed aromatic units during the torrefaction.12,23,25,27–34 | ||
An increasing intensity of the methoxyl group signal in lignin appeared after the thermal treatment of wood which is an enrichment of this functional group. The intensity of the CH2 carbons in the aliphatic chain (∼32 ppm) is almost intact after the torrefaction, which indicates that the decomposition of aliphatic chain (in fatty acids and tannins) at 250 °C is very limited. The methyl carbons in hemicellulose acetyl groups were no longer present after the torrefaction at 250 °C for 4 h, which is consistent with our previous results.
The CP/MAS 13C NMR spectra of torrefied wood samples produced by torrefaction of Loblolly pine wood at 250 and 300 °C are compared in Fig. 2. Significant differences of torrefied wood produced at the different temperatures were observed, for the same treatment time ∼30 min, the torrefied wood produced at 300 °C shows much higher intensities of carbonyl, carboxyl, aromatic carbons and methoxyl groups than the torrefied wood produced at 250 °C. In contrast, the methyl carbons in hemicellulose acetyl groups were almost completely decomposed. All of those results indicate that the decomposition reactions during the torrefaction were enhanced at higher reactor temperatures.4,7,8,11,17,18 The spectra shows that, after torrefaction at 300 °C for 4 h, the cellulose and hemicellulose in the wood were completely eliminated, the residue contains a large amount of carbonyl groups, aromatic carbons and methoxyl groups, which represent to the complex condensed aromatics, those aromatics were linked with aliphatic C–O bonds (60–100 ppm) and C–C bonds (10–20 ppm). A similar highly condensed aromatic polymer has been reported by Pastorova et al.13 They examined the structure of char produced by thermal treatment of cellulose at 190–390 °C and concluded that disproportionation occurred above ∼310 °C and lead to a highly condensed aromatic polymer. On the basis of literature reports,12,23,25,27–34 the possible pathway of decomposition of major wood components and the formation of new functional groups and condensed aromatics during the torrefaction is shown in Fig. 3.
Conclusions
The torrefaction of Loblolly pine wood was accomplished at 250 and 300 °C. The mass yields, HHVs and energy yields of torrefied wood are clearly impacted by the torrefaction temperature and time. Torrefaction at 250 °C for 4 h was found to be the optimal condition, which produced a torrefied wood that had a relatively high energy yield of 81.29% and a HHV of 24.06 MJ kg−1, which is comparable with several commercial coals.16 In addition, the carbohydrate analysis and CP/MAS 13C NMR spectra shows that the hemicellulose in the torrefied wood produced by torrefaction of wood at 250 °C for 4 h was almost completely decomposed, however, the cellulose and lignin were only slightly affected. Torrefaction of Loblolly pine at 300 °C for 4 h completely eliminated all the cellulose and hemicellulose in the original wood and the thermal residue had a complex aromatic structure and a very high HHV (32.34 MJ kg−1).Acknowledgements
The authors would like to acknowledge the financial support from the PSE Fellowship program at IPST@GT. This work is part of the first author's requirements for the degree of Ph.D. at Georgia Institute of Technology.Notes and references
- A. J. Ragauskas, C. K. Williams, B. H. Davison, G. Britovsek, J. Cairney, C. A. Eckert, W. J. Frederick, Jr., J. P. Hallett, D. J. Leak, C. L. Liotta, J. R. Mielenz, R. Murphy, R. Templer and T. Tschaplinski, Science, 2006, 311, 484–489 CrossRef.
- U. S. Energy Information Administration, International Energy Outlook 2010 http://www.eia.doe.gov/oiaf/ieo/emissions.html.
- K. David and A. J. Ragauskas, Energy Environ. Sci., 2010, 3, 1182 RSC.
- A. Pimchuai, A. Dutta and P. Basu, Energy Fuels, 2010, 24, 4638–4645 CrossRef CAS.
- R. Pentananunt, A. N. M. M. Rahman and S. C. Bhattacharya, Energy, 1990, 15, 1175–1179 CrossRef CAS.
- K. Svoboda, M. Pohořelý, M. Hartman and J. Martinec, Fuel Process. Technol., 2009, 90, 629–635 CrossRef CAS.
- M. Prins, K. Ptasinski and F. Janssen, J. Anal. Appl. Pyrolysis, 2006, 77, 28–34 CrossRef CAS.
- M. Prins, K. Ptasinski and F. Janssen, J. Anal. Appl. Pyrolysis, 2006, 77, 35–40 CrossRef CAS.
- W. Yan, T. C. Acharjee, C. J. Coronella and V. R. Vásquez, Environ. Prog. Sustainable Energy, 2009, 28, 435–440 Search PubMed.
- W.-H. Chen and P.-C. Kuo, Energy, 2010, 35, 2580–2586 CrossRef CAS.
- W.-H. Chen and P.-C. Kuo, Energy, 2011, 36, 803–811 CrossRef CAS.
- N. Brosse, R. El Hage, M. Chaouch, M. Pétrissans, S. Dumarçay and P. Gérardin, Polym. Degrad. Stab., 2010, 95, 1721–1726 CrossRef CAS.
- I. Pastorova, R. E. Botto, P. W. Arisz and J. J. Boon, Carbohydr. Res., 1994, 262, 27–47 CrossRef CAS.
- K. David, Y. Pu, M. Foston, J. Muzzy and A. Ragauskas, Energy Fuels, 2009, 23, 498–501 CrossRef CAS.
- Z. Hu, R. Sykes, M. F. Davis, E. Charles Brummer and A. J. Ragauskas, Bioresour. Technol., 2010, 101, 3253–3257 CrossRef CAS.
- S. A. Channiwala and P. P. Parikh, Fuel, 2002, 81, 1051–1063 CrossRef CAS.
- J. Deng, G.-j. Wang, J.-h. Kuang, Y.-l. Zhang and Y.-h. Luo, J. Anal. Appl. Pyrolysis, 2009, 86, 331–337 CrossRef CAS.
- H. Yang, R. Yan, H. Chen, D. Lee and C. Zheng, Fuel, 2007, 86, 1781–1788 CrossRef CAS.
- K. M. Holtman, N. Chen, M. A. Chappell, J. F. Kadla, L. Xu and J. Mao, J. Agric. Food Chem., 2010, 58, 9882–9892 CrossRef CAS.
- A. Pournou, Archaeometry, 2007, 50, 129–141.
- G. D. Love, C. E. Snape and M. C. Jarvis, Phytochemistry, 1998, 49, 1191–1194 CrossRef CAS.
- M. Dick-Pérez, Y. Zhang, J. Hayes, A. Salazar, O. A. Zabotina and M. Hong, Biochemistry, 2011, 50, 989–1000 CrossRef CAS.
- H. Ben and A. J. Ragauskas, Energy Fuels, 2011, 25, 2322–2332 CrossRef CAS.
- P. F. Britt, A. C. Buchanan, M. J. Cooney and D. R. Martineau, J. Org. Chem., 2000, 65, 1376–1389 CrossRef CAS.
- J. Zawadzki and M. Wisniewski, J. Anal. Appl. Pyrolysis, 2002, 62, 111–121 CrossRef CAS.
- S. Ates, M. H. Akyildiz and H. Ozdemir, BioResources, 2009, 4, 1032–1043 Search PubMed.
- T. Ahmad, R. Andersson, K. Olsson and O. Theander, Carbohydr. Res., 1993, 247, 211–215 CrossRef CAS.
- G. C. A. Luijkx, F. van Rantwijk and H. van Bekkum, Carbohydr. Res., 1993, 242, 131–139 CrossRef CAS.
- R. K. M. R. Kallury, C. Ambidge, T. T. Tidwell, D. G. B. Boocock, F. A. Agblevor and D. J. Stewart, Carbohydr. Res., 1986, 158, 253–261 CrossRef.
- D. K. Shen and S. Gu, Bioresour. Technol., 2009, 100, 6496–6504 CrossRef CAS.
- E. Jakab, K. Liu and H. L. C. Meuzelaar, Ind. Eng. Chem. Res., 1997, 36, 2087–2095 CrossRef CAS.
- F. Tao, H. Song and L. Chou, Carbohydr. Res., 2011, 346, 58–63 CrossRef CAS.
- F. Tao, H. Song and L. Chou, ChemSusChem, 2010, 3, 1298–1303 CrossRef CAS.
- J. Huang, C. Liu, S. Wei, X. Huang and H. Li, THEOCHEM, 2010, 958, 64–70 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |