Development of a fluorous, oxime-based palladacycle for microwave-promoted carbon–carbon coupling reactions in aqueous media†
Woen
Susanto
a,
Chi-Yuan
Chu
b,
Wei Jie
Ang
a,
Tzyy-Chao
Chou
b,
Lee-Chiang
Lo
*b and
Yulin
Lam
*a
aDepartment of Chemistry, National University of Singapore, 3 Science Drive 3, Singapore 117543. E-mail: chmlamyl@nus.edu.sg; Fax: 65-67791691; Tel: 65-65162688
bDepartment of Chemistry, National Taiwan University, No. 1, Sec. 4 Roosevelt Road, Taipei, 106, Taiwan. E-mail: lclo@ntu.edu.tw; Fax: 886-223636359; Tel: 886-233661669
First published on 14th November 2011
Abstract
A thermally stable, fluorous oxime-based palladacycle has been developed and was shown to efficiently promote various carbon–carbon bond formation reactions (Suzuki–Miyaura, Sonogashira and Stille) in aqueous media under microwave irradiation. The palladacycle gave extremely low levels of Pd leaching and could be reused five times with no significant loss of activity.
The palladium-catalyzed carbon–carbon coupling reaction is an important tool for the synthesis of symmetric and nonsymmetric biaryls and over the decades, a wide variety of homogeneous catalytic systems have been developed for this transformation.1 Although homogeneous catalysts have many advantages, the difficulties surrounding the separation and recovery of the catalyst, and product contamination with traces of heavy metals are common problems encountered, which restrict their applications in certain industries.2 This inherent limitation of homogeneous catalysis has thus resulted in the development of new strategies for transition-metal catalysis that facilitates catalyst recovery and recycling.3,4 One of these strategies which has attracted much interest involves fluorous chemistry.4 However, these methods of catalysis are typically conducted in organic or perfluorocarbon solvents.5 From the environmentally benign viewpoint, the use of water as a solvent in transition-metal catalysis offers many advantages.6 Although various catalytic systems in aqueous solutions have been developed,7 very few examples of fluorous palladium-based catalysis in aqueous media have been reported.8 In 2008, a recyclable polymer-supported fluorous catalyst (Amberlyst A-21-Pd(OPf)2, 1 mol% Pd) was shown to promote the Sonogashira reaction in water at 80 °C in 1–18 h (Pd leaching: 3.6–11.6 ppm)8a and recently, recyclable fluoro silica gel-supported perfluoro-tagged palladium nanoparticles (0.1 mol% Pd) were used to catalyze the Suzuki–Miyaura reaction in water at 100 °C in 8–12 h (Pd leaching: <10 ppm).8b In 2009, Theberge et al. have also reported a fluorous–aqueous biphasic Suzuki–Miyaura coupling in droplets at room temperature in 0.75–8 h (Pd leaching: <0.5 ppm).8c Herein we report the synthesis of a recyclable, fluorous, oxime-based palladacycle 1 (Scheme 1) and illustrate its application in the various carbon–carbon coupling reactions under aqueous conditions and microwave irradiation. An oxime-derived palladacycle was chosen because it is known to be (i) thermally robust, (ii) insensitive to air and moisture, (iii) a precatalyst with a high ability to slowly release Pd(0) which suppresses the growth of large Pd metal particles,9 and (iv) phosphines are not involved in the reaction and thus problems involving phosphine oxide and side-reactions arising from the coupling between phosphine and aryl halide would be avoided.10
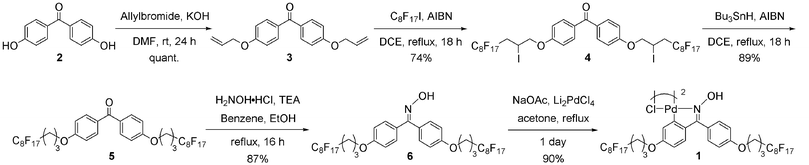 | ||
| Scheme 1 Synthesis of palladacycle 1. | ||
Palladacycle 1 was prepared by treating 4,4′-dihydroxybenzophenone 2 with allyl bromide at room temperature for a day to give the allyl ether 3 in quantitative yield. Compound 3 was then reacted with perfluorooctyl iodide (C8F17I) in the presence of azobisisobutyronitrile (AIBN) in a radical reaction to give compound 4 in 74% yield. Removal of the iodide in compound 4 was carried out using tributyltin hydride (Bu3SnH) and AIBN in dichloroethane at refluxing temperature to give compound 5 in 89% yield. Subsequent condensation of 5 with hydroxylamine hydrochloride (H2NOH·HCl) gave oxime 6 (87% yield) which in turn was treated with tetrachlorolithiumpalladate (Li2PdCl4) in the presence of a base to produce palladacycle 1.
For the synthesis of palladacycle 1, we had initially used the reaction conditions reported by Alacid and Nájera.11 However, formation of Pd black was observed during the reaction and palladacycle 1 was obtained in low yield (Table S1 in the ESI†, entry 1). This could be attributed to the insolubility of compound 6 in methanol. In order to optimize the reaction, we varied the solvent and reaction time and found that the best result was obtained in acetone. However at room temperature, the reaction required 6 days to complete. This long reaction time could be due to the poor solubility of sodium acetate in acetone. Hence, we carried out the reaction under reflux and indeed, the reaction time was shortened to 1 day and palladacycle 1 was obtained in 90% yield (Table S1 in ESI†, entry 4).
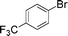
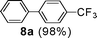
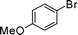
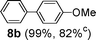
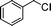
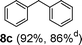
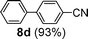
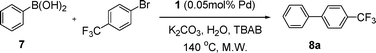
Initial assessment of the catalytic activity of palladacycle 1 (0.05 mol% Pd) was conducted using the Suzuki–Miyaura reaction between phenylboronic acid 7 and 4-bromobenzotrifluoride with K2CO3 as the base, water as the solvent, TBAB as an additive and under microwave irradiation at 140 °C. The reaction proceeded efficiently to give the biphenyl product 8a in 98% yield within 2 min (Table 1, entry 1). It is worth noting that other solvent systems, including THF which is commonly used for the homogeneous palladium-catalyzed Suzuki–Miyaura reaction, gave lower yields of the product (Table S2†, entries 3–6). We next examined the effects of the bases on the reaction. Two inorganic bases (K2CO3 and KOH) and an organic base (Cy2NMe) were investigated, and K2CO3 was found to be the most effective base for the reaction (Table S2†, entries 1, 7–8). The reaction temperature was also varied and the experimental data obtained (Table S2†, entries 1, 9–10) indicated that although the reaction completed more rapidly under microwave irradiation conditions, raising the temperature to 170 °C resulted in a lower yield of the product. Finally, we tried to reduce the precatalyst loading to 0.005 mol% Pd. This afforded 8a in 98% yield (corresponding to 2 × 104 TON) and the reaction time was only slightly longer than the reaction with 0.05 mol% Pd (Table S2†, entries 1 and 2). However, when 0.005 mol% Pd was applied to the cross-coupling of 7 with benzyl chloride under microwave irradiation at 140 °C, the reaction required nearly an hour to complete. Thus, a precatalyst loading of 0.05 mol% Pd was used for the remaining Suzuki–Miyaura reactions with palladacycle 1 (Table 1). To determine the catalytically active species in the reaction, we carried out the mercury drop test and another test using substoichiometric amounts of CS2. Results from both tests (Table S2†, entries 11 and 12) strongly suggest that the palladium nanoparticles are the catalytic species in the reaction. A comparison of palladacycle 1 with its polymer-supported analog showed that the Suzuki–Miyaura reaction with palladacycle 1 occurred more rapidly and gave a higher yield of the product (Table 1, entries 2–3). It was also gratifying to note that our reaction protocol could be applied successfully to the synthesis of diarylmethanes (Table 1, entry 3), as such syntheses have been reported to be difficult using various homogeneous palladium catalyst systems12 and was only recently made more accessible viapalladium–phosphine catalysts.13
| Entry | ArX | Time (min) | Product (Yieldb) |
|---|---|---|---|
a Palladacycle 1 (0.05 mol% Pd), K2CO3, TBAB, H2O, 140 °C, M.W.
b Isolated yield.
c Using polymer-supported oxime-based palladacycle:1 0.1 mol% Pd, K2CO3, H2O, 100 °C.
d Using polymer-supported oxime-based palladacycle:1 0.1 mol% Pd, KOH, TBAB, acetone–H2O (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2), 50 °C. :2), 50 °C.
|
|||
| 1 |
|
2 |
|
| 2 |
|
6 (2.5 hc) |
|
| 3 |
|
3.5 (1 hd) |
|
| 4 |
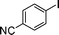
|
2 |
|
Next, we investigated the possibility of recovering and reusing palladacycle 1. 4-Bromobenzotrifluoride and 7 were used for the model study under the optimized reaction conditions. The recycling experiments were carried out over 5 runs and the time taken for the reaction to reach completion was 2–7 min to produce 8a in 92–98% yields (Table 2). In addition, ICP-OES analysis of Pd leaching in the crude product was very low (0.023–0.033 ppm over 5 cycles). It is worth noting that the amount of palladium leaching is much much lower than that observed for the polymer-supported oxime-based palladacycle analog (14.4–20.2 ppm over 7 cycles)14 and the three earlier reported fluorous Pd-catalysts which were applied to the cross-coupling reactions in aqueous media (vide supra).8
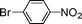
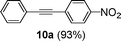
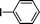
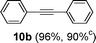
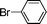
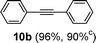
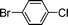
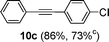
Encouraged by these results, we extended our studies to the Stille and copper-free Sonogashira coupling reactions. Attempts to carry out the Sonogashira coupling of p-nitrophenyl bromide and phenylacetylene 9 with palladacycle 1 (0.05 mol% Pd) and different bases under microwave irradiation at 140 °C resulted in a sluggish reaction (Table S3†, entries 1, 3–5). Hence the precatalyst loading was increased to 0.5 mol% Pd and the best yield was obtained when pyrrolidine was used as the base (Table S3†, entries 1–5). Next, we varied the solvent and found that water proved to be the best solvent (Table S3†, entries 5–9). These results were gratifying as Sonogashira coupling reactions in neat water and in the absence of additives15 are rare and low yielding because of the poor solubility or instability of the catalysts and coupling reagents in aqueous media.16 For our experiments into the Sonogashira coupling reaction in aqueous media, we had initially used undegassed water for the reaction. This resulted in the homocoupled product of 9 being formed as a side-product (Table S3†, entry 9). To circumvent this problem, we tried degassed water and found that not only was the homocoupled product absent but the reaction time was also reduced. To demonstrate the versatility of palladacycle 1 for copper-free Sonogashira coupling reactions, we have applied it to other substrates which also gave good yields of the desired product (Table 3).
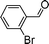
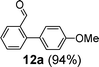
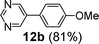
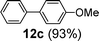
Next, we investigated the applicability of palladacycle 1 to the Stille coupling reaction. In our initial studies, palladacycle 1 (0.05 mol% Pd) was added to tributyl(4-methoxyphenyl)stannane 1118 and o-bromobenzaldehyde in different solvents (Table S4 in the ESI†, entries 1–4). The best yield was obtained when water was used as the reaction solvent but the reaction time was 28 min. To reduce the reaction time, we tried increasing the reaction temperature but this gave a poorer yield of the desired product. Thus we introduced TBAB as an additive and this resulted in a higher yield (91%) and a significant reduction in reaction time (2 min) (Table S4†, entry 6). Further experimentation showed that the reaction proceeded equally well at a lower temperature (100 °C) and precatalyst loading (0.005 mol% Pd), giving the product in both a comparable yield and reaction time (Table S4†, entry 8). Using these optimized conditions, we have extended the study to other substrates which also gave good to excellent yields of the desired products (Table 4).
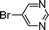
Conclusion
In summary, a fluorous oxime-based palladacycle 1 was synthesized and shown to promote the Suzuki–Miyaura, Sonogashira and Stille coupling reactions in aqueous media. Palladacycle 1 could be used under microwave irradiation at high temperatures which significantly shortened the reaction time. Recycling was also possible with palladacycle 1 which was reused five times in a Suzuki–Miyaura reaction without significant loss of activity. Palladium leaching was also very low.Acknowledgements
This research is supported by grants from the National University of Singapore (R-143-399-112 to Y. L.) and National Science Council of Taiwan (NSC 98-2113-M-002-009-MY3 to L.-C.L.).Notes and references
- (a) J. Tsuji, Palladium Reagents and Catalyst: Innovations in Organic Synthesis, 1996, John Wiley & Sons Ltd, Chichester, England Search PubMed; (b) T. Hayashi, J. Organomet. Chem., 2002, 653, 41 CrossRef CAS; (c) I. Moreno, R. San Martin, M. T. Herrero and E. Dominguez, Curr. Top. Catal., 2009, 8, 91 CAS; (d) J. Dupont, F. R. Flores, Handbook of Green Chemistry, 2009, Wiley-VCH, Weinheim, Germany, pp 319 Search PubMed.
- (a) E. B. Mubofu, J. H. Clark and D. Macquarrie, Green Chem., 2001, 3, 23 RSC; (b) D. Rosario-Amorin, X. Wang, M. Gaboyard, R. Clérac, S. Nlate and K. Heuzé, Chem.–Eur. J., 2009, 15, 12636 CrossRef CAS.
- (a) N. E. Leadbeater and M. Marco, Chem. Rev., 2002, 102, 3217 CrossRef CAS; (b) T. J. Dickerson, N. N. Reed and K. D. Janda, Chem. Rev., 2002, 102, 3325 CrossRef CAS; (c) Q.-H. Fan, Y.-M. Li and A. S. C. Chan, Chem. Rev., 2002, 102, 3385 CrossRef CAS; (d) A. Molnár, Chem. Rev., 2011, 111, 2251 CrossRef.
- I. T. Horváth and J. Rábai, Science, 1994, 266, 72 Search PubMed.
- N. Audic, P. W. Dyer, E. G. Hope, A. M. Stuart and S. Suhard, Adv. Synth. Catal., 2010, 352, 2241 CrossRef CAS and references therein.
- C.-J. Li, Chem. Rev., 2005, 105, 3095 CrossRef CAS.
- (a) G. Hamasaka, T. Muto and Y. Uozumi, Angew. Chem., Int. Ed., 2011, 50, 4876 CrossRef CAS; (b) M. Lamblin, L. Nassar-Hardy, J.-C. Hierso, E. Fouquet and F.-X. Felpin, Adv. Synth. Catal., 2010, 352, 33 CrossRef CAS; (c) Y. Uozumi and Y. M. A. Yamada, Chem. Rec., 2009, 9, 51 CrossRef CAS; (d) K. H. Shaughnessy, Chem. Rev., 2009, 109, 643 CrossRef CAS.
- (a) Z.-W. Ye and W.-B. Yi, J. Fluorine Chem., 2008, 129, 1124 CrossRef CAS; (b) L. Wang and C. Cai, J. Mol. Catal. A: Chem., 2009, 306, 97 CrossRef CAS; (c) A. B. Theberge, G. Whyte, M. frensel, L. M. Fidalgo, R. C. R. Wootton and W. T. S. Huck, Chem. Commun., 2009, 6225 RSC.
- N. T. S. Phan, M. Van Der Sluys and C. W. Jones, Adv. Synth. Catal., 2006, 348, 609 CrossRef CAS.
- (a) I. P. Beletskaya and A. V. Cheprakov, J. Organomet. Chem., 2004, 689, 4055 CrossRef CAS; (b) D. A. Alonso, C. Nájera and M. C. Pacheco, Org. Lett., 2000, 2, 1823 CrossRef CAS.
- E. Alacid and C. Nájera, Adv. Synth. Catal., 2007, 349, 2572 CrossRef CAS.
- (a) L. Botella and C. Nájera, J. Organomet. Chem., 2002, 663, 46 CrossRef CAS; (b) D. A. Alonso, C. Nájera and M. C. Pacheco, J. Org. Chem., 2002, 67, 5588 CrossRef CAS.
- (a) N. Henry, C. Enguehard-Gueiffier, I. Thery and A. Gueiffier, Eur. J. Org. Chem., 2008, 28, 4824 CrossRef; (b) I. A. Fairlamb, P. Sehnal and R. J. K. Taylor, Synthesis, 2008, 2009, 508 CrossRef.
- E. Alacid and C. Nájera, J. Organomet. Chem., 2008, 694, 1658 CrossRef.
- Additives are added to solubilize the reagents and catalysts: D. Rosario-Amorin, M. Gaboyard, R. Clérac, S. Nlate and K. Heuze, Dalton Trans., 2011, 40, 44 RSC and references therein.
- (a) B. Liang, M. Dai, J. Chen and Z. Yang, J. Org. Chem., 2005, 70, 391 CrossRef CAS; (b) B. Inés, R. San Martin, F. Churruca, E. Dominguez, M. K. Urtiaga and M. I. Arriortua, Organometallics, 2008, 27, 2833 CrossRef.
- F. Habib, I. Nasser and G. Mohammad, J. Mol. Catal. A: Chem., 2010, 321, 110 CrossRef.
- Synthesis of 11: L. S. Liebeskind and J. Wang, Tetrahedron, 1993, 49, 5461 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, Tables S1–S4 and full spectroscopic data for all new compounds. See DOI: 10.1039/c1gc16108c |
| This journal is © The Royal Society of Chemistry 2012 |