Electrostatic immobilization of an olefin metathesis pre-catalyst on iron oxide magnetic particles
Matthew J.
Byrnes
,
Andrew M.
Hilton
,
Clint P.
Woodward
,
William R.
Jackson
and
Andrea J.
Robinson
*
School of Chemistry, Monash University, Clayton, 3800, Australia. E-mail: andrea.robinson@monash.edu; Fax: +61 3 9905 4597; Tel: +61 3 9905 4553
First published on 15th November 2011
Abstract
A quaternary ammonium Hoveyda-Grubbs olefin metathesis pre-catalyst has been reversibly immobilized on sulphonic acid-functionalised silica-coated iron oxide magnetic particles to affect ring closing metathesis with easy removal, reuse and regeneration.
Introduction
Homogenous Grubbs-type pre-catalysts have played an important role in the application of olefin metathesis to organic synthesis.1,2 Grafting of these homogeneous catalysts onto solid supports can aid catalyst recycling and removal of metallic residues from the product stream by filtration.3 This strategy, however, must take into consideration the effect of the solid support on catalytic efficiencies. Immobilization of the catalysts onto polymeric supports, for example, can often result in lower activity due to poorer substrate diffusion.4Silica supports, on the other hand, are not subject to the same physiochemical constraints (i.e.catalyst-polymer solvation) and the availability of a large surface area for functionalisation aids the activity of the final immobilized catalyst.5 However, final recovery of silica-based catalysts from the reaction stream can still be problematic. More recently, magnetic particles (MPs) have been used as solid supports to conduct ‘pseudo-homogenous’ transition-metal catalysed reactions.6 The application of an external magnet readily separates the catalyst-loaded MPs from the reaction products to eliminate the need for filtration and facilitate catalyst recycling.7Towards this end, palladium, osmium and nickel catalysts have been attached to MPs,8–12 and more recently, two examples of covalent attachment of Hoveyda-Grubbs metathesis catalysts to MPs have been reported.13,14Catalyst-tethering strategies employed in the metathesis area to date largely involve immobilization of the Ru-alkylidene catalystsvia covalent attachment to the neutral ligand (either phosphine or N-heterocyclic carbene), the benzylidene or a chloride surrogate.15,16 A few examples of immobilisation via non-covalent modes have also been reported and these include the use of π–π stacking and electrostatic interactions.17,18
In this study the use of reversible electrostatic immobilization of Hoveyda-Grubbs metathesis catalysts to sulphonated iron oxide MPs has been investigated. The quartenary ammonium-functionalised Ru-benzylidene pre-catalyst (1),19 was readily attached to sulphonated MPs to generate a high functioning and reloadable metathesis catalyst. Conveniently, the MP-catalyst was easily magnetically retrieved and reused in subsequent catalysis experiments and also readily recharged with fresh pre-catalyst to maintain optimum performance (turn-over number and frequency).
Results and discussion
Iron oxide magnetic particles with a shell of silica were prepared via a co-precipitation reaction. The sulphonic acid groups were installed by reacting 3-mercaptotrimethoxysilane with freshly prepared Si-coated Fe3O4 MPs followed by oxidation of the mercapto groups with 30% H2O2. The amount of sulphonic acid present on the particles, as determined by titration with 0.1 M NaOH, ranged from 0.49 to 0.51 mmol g−1. Recrystallised catalyst 1 was successfully immobilised onto the sodium sulphonated iron oxide particles in dry, degassed CH2Cl2 to form the MP-appended pre-catalyst 2 as shown in Scheme 1. ICP-MS ruthenium analysis of the pre-catalyst 2 found that catalyst loading was lower than that anticipated, with a value of 0.12 mmol g−1, possibly indicating a surface crowding effect and/or inaccessible sulphonate groups. A SEM image of particles of 2 revealed that many of the particles had nanosized structure (<100 nm).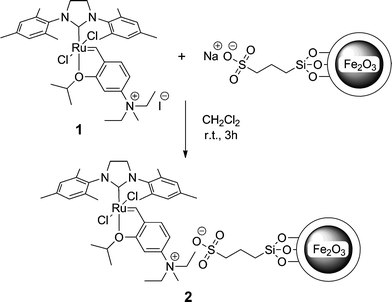 | ||
| Scheme 1 | ||
Ring closing metathesis (RCM) of diethyl diallylmalonate (3) was chosen to evaluate the immobilized catalyst (Scheme 2). A trial run using 0.68 mol% loading of 2 on substrate 3 showed >95% conversion to the cyclopentene 4 after 2 hours (Table 1) and gave a clear solution after magnetic separation of the catalyst particles. Comparable conversion of 3 into 4 was also obtained with catalyst 1, however removal of the ruthenium-alkylidene catalyst and its decomposition products from the ring-closed product 4via organic-aqueous phase extraction and chromatography was not as effective. A visual comparison of the effectiveness of catalyst separation can be seen in Figure 1.
| Cycle | Time (h) | Method (% conversion)a | Ru (ppm)b | |||
|---|---|---|---|---|---|---|
| A | B | C | D | 4 | ||
| Reactions run with 0.68 mol% catalyst loading and triplicate washing. Methods A and B: RCM performed with 5 mL of 0.047 M 3 at room temperature and reaction times of 2 h (Cycles 1–5) and 12 h (Cycle 6). Method A uses DCM and Method B uses toluene. Methods C and D: RCM performed with a nitrogen bleed using 5 mL of 0.047 M 3 at room temperature and reaction times of 2 h (Cycles 1–5) and 12 h (Cycle 6). Method C uses DCM and Method D uses toluene. n.d. = not determined.a Conversions to 4 calculated by 1H NMR spectroscopy.b Residual ruthenium analysis in cyclised product 4 generated using Method D. | ||||||
| 1 | 2 | >95 | >95 | >95 | >95 | 200 |
| 2 | 2 | >95 | >95 | >95 | >95 | 114 |
| 3 | 2 | 84 | 90 | >95 | >95 | 100 |
| 4 | 2 | 53 | 83 | 86 | >95 | 62 |
| 5 | 2 | 56 | 71 | 81 | 87 | 43 |
| 6 | 12 | 87 | >95 | n.d. | n.d | |
 | ||
| Fig. 1 NMR solutions of organics isolated from the RCM of 3. Shown on the left is the solution after magnetic separation of the MP-immobilised catalyst 2. The retrieved catalyst was recycled in subsequent reactions. The solution on the right has not been treated to remove Ru-containing residues and the catalyst 1 was not recovered. | ||
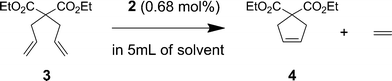 | ||
| Scheme 2 | ||
To test the recovery and reuse of the MP catalyst system, a series of recycling experiments was conducted over 5 cycles (washing 3 × 5 mL of solvent between each cycle) (Table 1) keeping the reaction time constant.
The RCM of 3 was found to proceed smoothly with >95% conversion over the first two cycles (Cycles 1 and 2) for reactions performed in both CH2Cl2 (Method A) and toluene (Method B). However, a decrease in conversion was experienced after the second cycle (Cycles 3–5, Methods A and B). The reduction in conversion was less pronounced when performed in toluene. High conversion (>80%), however, was reachieved with a longer reaction time (12 h) in both cases (Cycle 6). In addition to producing superior conversion, the use of toluene also aided magnetic recovery by promoting particle aggregation once magnetic stirring had ceased.
Introduction of a bleed needle to purge ethylene byproduct during reaction proved to be beneficial in both solvents over 5 consecutive runs (Table 1, Cycles 1–5, Methods C and D). In toluene, high conversions (above 87%) were maintained over 5 consecutive cycles. In each cycle, solvent volume remained constant over the 2 h reaction period, and high conversion to 4 was observed in both CH2Cl2 and toluene.
We considered that the observed decline of catalyst activity over five runs could be explained in a number of ways: Physical loss of the catalyst-MP construct, catalyst deactivation, or ruthenium alkylidene leeching from the MPs. Gradual loss of magnetic particles through failure to recover the MPs at the end of each catalytic cycle would contribute to a decline in reactivity. The use of a high strength recovery magnet (0.45T) minimised this problem, however fine particulate material, possibly resulting from mechanical degradation of the MPs during stirring, could not be recovered. Catalyst loss via this route, however, was considered to be minimal.
The stability of the catalyst system to oxygen and water was then assessed. The use of degassed toluene which had not been subjected to a drying procedure (i.e. used as supplied) did not compromise catalyst reactivity (all conversions >95%), whereas non-degassed, dry toluene significantly affected the performance of the catalyst. Exposure of dried particles to atmospheric conditions also compromised catalyst lifetime. It is therefore important to protect the MP-catalyst system from oxygen exposure by storing under an inert atmosphere at all times after preparation. ICP-MS analysis of recovered 2 and isolated samples of 4 from successive cycles revealed that the Ru-loading on the MPs decreased with each successive cycle. Furthermore, analysis of the organic samples showed significant quantities of ruthenium ranging from 200 ppm for the first cycle product to 43 ppm for the final product isolated from cycle 5 (Table 1). Analysis of the 5th cycle MP-catalyst 3 indicated a loading of 0.06 mmol g−1 of ruthenium, a loss of ∼50% of the initial ruthenium content.
The loss of ruthenium and catalyst activity after only 4 cycles under optimum reaction conditions (Method D) was perplexing given that Jiang and coworkers, using a structurally related, covalently ligated Hoveyda-Grubbs-MP catalyst construct, were able to perform 13 sequential RCM reactions in near quantitative yield.13 While loss of MP-bound Ru could arise from separation of the pre-catalyst 1 from the MP at the ionic attachment site, the use of rigorously dried solvents protects the catalyst construct from dissociation. Instead we believe that ineffective recapture of in situ generated homogeneous Ru-alkylidenes is the major cause of ruthenium leeching from the MPs.20 Significantly, Plenio and coworkers have recently shown that the release-return mechanism does not play a significant role in catalytic RCM cycles employing Hoveyda-Grubbs-type catalysts.21 Hence, in general, immobilisation strategies via the labile benzylidene ligand are likely to result in sub-optimum performance. The initial value of 200 ppm in product 4 after cycle 1 (Table 1), however, is comparable to several other reported trace residual Ru values obtained after a variety of metal-removal treatments.22
Once it was identified that the activity of the catalyst was decreasing over time we chose to investigate whether the sub-performing particles could be regenerated. The particles were stripped of remaining catalyst by washing with 1 M HCl. This process disrupted the electrostatic interaction between the quaternised ligand and the particles. After washing with additional portions of water and thorough drying, the particles were retitrated with 0.1 M NaOH, giving a concordant value of 0.44 mmol g−1, slightly lower than the initial preparation. ICP-MS ruthenium analysis of the stripped MPs revealed a residual ruthenium content of 0.018 mmol g−1. Attempted RCM of 3 with these particles gave no conversion to 4 indicating that only non-catalytically active ruthenium species and/or non-accessible 2 was bound to the MP.
A fresh stoichiometric portion of catalyst 1 was then added to the recovered MPs as previously described. ICP-MS analysis of the regenerated Ru-magnetic particles gave a Ru analysis value of 0.075 mmol g−1 which was lower than the original MP-pre-catalyst value of 0.12 mmol g−1. Additional catalysis runs were performed with the reduced catalyst loading (adjusted to 0.43 mol% in contrast to the previously employed 0.68 mol% loading shown in Table 1) and >95% yields for the RCM of 3 into 4 were obtained over two successive reaction cycles using the regenerated catalyst.
Conclusion
A quaternary ammonium Hoveyda-Grubbs olefin metathesis pre-catalyst 1, which is readily accessed in one step from commercially available second generation Grubbs catalyst, has been successfully immobilized onto magnetically separable nanosized iron oxide particles. The resultant ruthenium alkylidene catalyst provided pseudo-homogeneous reactivity coupled with an in-built facile recovery option. The use of electrostatic attachment also enabled ready reloading of the catalyst and reuse of the functionalised MPs. Magnetic retrieval of the immobilised catalyst simplified product isolation and catalysis recycling.Experimental
General
All chemicals and solvents were used as purchased with the following exceptions. Toluene was distilled over Na wire and argon sparged prior to use. Dichloromethane was distilled over CaH2 and argon sparged prior to use. THF was distilled from benzophenone and Na wire prior to use. TEOS refers to tetraethoxysilane. Quaternary ammonium catalyst 1 was prepared according to a literature method19 and recrystallised from dichloromethane/hexane.1H NMR spectra were acquired using a Bruker DPX 300 MHz spectrometer (300 MHz 1H) or a Bruker DRX 400 MHz spectrometer (400 MHz 1H) as solutions in CDCl3. Chemical shifts (δ) were calibrated against the residual solvent peak.
ICP-MS analyses were conducted using the a previously reported procedure23 by Dr Ian McDonald of Earth and Ocean Sciences, Cardiff University.
Preparation of sulphonic acid functionalised iron oxide nanoparticles
A combined solution of FeCl3.6H2O (8.63 mL, 1 M) and FeCl2·4H2O (2.16 mL in 2 M HCl) was added to a rapidly stirred solution of aqueous NH3 (281 mL, 0.7 M). A dark brown precipitate of Fe3O4 formed instantly. The mixture was stirred for 1 h before being magnetically separated. The bulk of the solution was decanted from the particles, and the resultant slurry of the magnetic particles (in 25 mL) was combined with water (25 mL), EtOH (175 mL) and NH3 (1.3 mL, 28%) before dropwise addition of TEOS (1.3 g). The resulting mixture was left to stir overnight. The ethanol was removed from the aqueous solution by rotary evaporation and MeOH (500 mL) and NH3 (1.3 mL, 28%) added. 3-Mercaptopropyltrimethoxysilane (2.6 g, 0.013 mol) was then added and the mixture was left to stir for 3 days at room temperature. The particles were then magnetically separated and the resulting slurry was washed with water (500 mL) before the addition of H2O2 (30 mL, 30%). This solution was stirred overnight and then separated by centrifugation. After the particles had been isolated they were washed with 1 M HCl (25 mL) and water (2 × 100 mL) before the remaining solvent was removed by rotary evaporation. The isolated particles were then dried under high vacuum (25–30% yield).Titration of sulphonic acid functionalised iron oxide MPs
Sulphonic acid functionalised particles (239 mg) were sonicated in brine (10 mL) until a homogeneous slurry was obtained. The slurry was then titrated against NaOH (0.1 M) to neutral pH (determined by a pH meter). The particles were then washed with water (20 mL), reacidified by exposure to HCl (20 mL, 1 M) for 30 minutes, and then again washed with water (3 × 20 mL). This process was repeated until concordant titration readings were obtained. The titrated particles were stored in a glovebox and used for catalyst adhesion after drying under high vacuum.Preparation of 2
Dry, deoxygenated CH2Cl2 (10 ml) was added via syringe to a nitrogen purged Schlenk flask containing 1 (25 mg, 0.029 mmol) and 50 mg of the ca. 0.5 mmol g−1sodium sulfonated functionalised iron oxide magnetic particles (ca. 0.025 mmol). The solution was stirred at room temperature for 3 h. The particles were then magnetically separated, washed with dry, degassed CH2Cl2 (5 × 10 mL each), dried under high vacuum and stored in a glovebox prior to use.RCM assessment of 2
Diethyl diallyl malonate 3 (5 mL of a 0.047 M stock solution in dry degassed solvent) was added to an argon purged Schlenk flask containing 2 (13.3 mg, ∼0.68 mol%) and the resulting solution was stirred at room temperature for 2 h. The particles were magnetically separated over a five minute period and then washed with the designated solvent (3 × 10 ml). This cycle was repeated 4 times with addition of fresh substrate (5 mL of a 0.047 M stock solution in dry degassed solvent). Both dichloromethane and toluene were used as solvents. The use of a bleed needle was employed where stated.Regeneration of the catalyst particles
Spent catalyst particles 2 (70 mg) were washed with water (50 mL) before being dispersed in 1 M HCl (25 mL) and stirred for 30 min at room temperature. The acid treated particles were then washed with water (2 × 50 mL) and dried under high vacuum. The cleaved particles were retitrated using the same protocol as stated previously, giving a titration value of 0.44 mmol g−1. After titration the particles were washed with water (3 × 20 mL) and THF (1 × 20 mL), then dried under high vacuum.Regeneration with catalyst 1 was performed by adding dry, deoxygenated dichloromethane (5 ml) to a nitrogen-purged Schlenk flask containing 1 (6.3 mg, 8.83 μmol) and 20 mg of 0.44 mmol g−1sodium sulfonated functionalised iron oxide magnetic particles (ca. 0.025 mmol). The solution was stirred at room temperature for 3 h. The particles were then magnetically separated, washed with dry, degassed dichloromethane (5 × 10 mL), dried under high vacuum and stored in a glovebox prior to use.
Acknowledgements
This work was partially supported by an Early-Career Reseacher Grant to MJB by the Faculty of Science, Monash University, Australia. ICP-MS analyses were performed by Dr Ian McDonald (Earth and Ocean Sciences, Cardiff University) with the generous assistance of Prof. David Knight.References
- A. H. Hoveyda and A. R. Zhugralin, Nature, 2007, 450, 243–251 CrossRef CAS.
- P. H. Deshmukh and S. Blechert, Dalton Trans., 2007, 2479–2491 RSC.
- H. Clavier, K. Grela, A. Kirschning, M. Mauduit and S. P. Nolan, Angew. Chem., Int. Ed., 2007, 46, 6786–6801 CrossRef CAS.
- S. T. Nguyen and R. H. Grubbs, J. Organomet. Chem., 1995, 497, 195–200 CrossRef CAS.
- D. P. Allen, M. M. Van Wingerden and R. H. Grubbs, Org. Lett., 2009, 11, 1261–1264 CrossRef CAS.
- (a) A. H. Lu, L. Salabas and F. Schuth, Angew. Chem., Int. Ed., 2007, 46, 1222–1244 CrossRef CAS; (b) V. Polshettiwar, R. Luque, A. Fihri, H. Zhu, M. Bouhrara and J. M. Basset, Chem. Rev., 2011, 111, 3036–3075 CrossRef CAS.
- (a) P. D. Stevens, G. Li, J. Fan, M. Yen and Y. Gao, Chem. Commun., 2005, 4435–4437 RSC; (b) M. Shokouhimehr, Y. Piao, J. Kim, Y. Jang and T. Hyeon, Angew. Chem., Int. Ed., 2007, 46, 7039–7043 CrossRef CAS.
- K-I. Fujita, S. Umeki, M. Yamazaki, T. Ainoya, T. Tsuchimoto and H. Yasuda, Tetrahedron Lett., 2011, 52, 3137–3140 CrossRef CAS.
- J. Liu, X. Peng, W. Sun, Y. Zhao and C. Xia, Org. Lett., 2008, 10, 3933–3936 CrossRef CAS.
- V. Polshettiwar and R. S. Varma, Chem.–Eur. J., 2009, 15, 1582–1586 CrossRef CAS.
- T. Hirakawa, S. Tanaka, N. Usuki, H. Kanzaki, M. Kishimoto and M. Kitamura, Eur. J. Org. Chem., 2009, 789–792 CrossRef CAS.
- R. Abu-Reziq, D. Wang, M. Post and H. Alper, Chem. Mater., 2008, 20, 2544–2550 CrossRef CAS.
- C. Che, W. Li, S. Lin, J. Chen, J. Zheng, C-C. Wu, Q. Zheng, G. Zhang, Z. Yang and B. Jiang, Chem. Commun., 2009, 5990–5992 RSC.
- Z. Yinghuai, L. Kuijin, N. Huimin, L. Chuanzhao, L. P. Stubbs, C. F. Siong, T. Muihua and S. C. Peng, Adv. Synth. Catal., 2009, 351, 2650–2656 CrossRef.
- M. R. Buchmeiser, New J. Chem., 2004, 28, 549–557 RSC.
- C. Coperet and J. M. Basset, Adv. Synth. Catal., 2007, 349, 78–92 CrossRef CAS.
- G. Liu, B. Wu, J. Zhang, X. Wang, M. Shao and J. Wang, Inorg. Chem., 2009, 48, 2383–2390 CrossRef CAS.
- A. Michrowska, K. Mennecke, U. Kunz, A. Kirschning and K. Grela, J. Am. Chem. Soc., 2006, 128, 13261–13267 CrossRef CAS.
- A. Michrowska, L. Gulajski, Z. Kaczmarska, K. Mennecke, A. Kirschning and K. Grela, Green Chem., 2006, 8, 685–688 RSC.
- D. Rix, F. Caijo, I. Laurent, L. Gulajski, K. Grela and M. Mauduit, Chem. Commun., 2007, 3771–3773 RSC.
- T. Vorfalt, K. J. Wannowius, V. Thiel and H. Plenio, Chem.–Eur. J., 2010, 16, 12312–12315 CrossRef CAS.
- G. C. Vougioukalakis and R. H. Grubbs, Chem. Rev., 2010, 110, 1746–1787 CrossRef CAS.
- D. W. Knight, I. R. Morgan and A. J. Proctor, Tetrahedron Lett., 2010, 51, 638–640 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |