Liquid hydrocarbon fuels from cellulosic feedstocks via thermal deoxygenation of levulinic acid and formic acid salt mixtures
Paige A.
Case
,
Adriaan R. P.
van Heiningen
and
M. Clayton
Wheeler
*
Department of Chemical and Biological Engineering and Forest, Bioproducts Research Institute, 5737 Jenness Hall, Orono, ME 04469-5737, USA. E-mail: cwheeler@umche.maine.edu; Fax: +1 207 581 2323; Tel: +1 207 581 2280
First published on 18th November 2011
Abstract
Formic acid is demonstrated as a hydrogen source in a solid reaction system by first stabilizing the acid as a calcium salt which then decomposes at temperatures of relevance in pyrolytic reactions. High yields of deoxygenated hydrocarbons are produced by thermal decomposition of formic and levulinic acid mixtures where the optimum feed stoichiometry is consistent with that of cellulose hydrolysis and dehydration. The method promises a high-yield, robust, low-pressure, non-catalytic route for converting biomass hydrolyzates to hydrocarbon mixtures which are similar to petroleum crude oils.
Two techno-economic barriers hinder commercial implementation of renewable transportation fuel production from lignocellulosic biomass. First, the capital cost per unit product is high because the present largest wood processing facilities, as exemplified by pulp mills, are almost two orders of magnitude smaller in feed stock energy feed rate than an average oil refinery. The worlds largest pulp mills are capable of processing about 2 million dry metric tons (MT) per year,1 and a large refinery can process over 200 million barrels per year.2 Therefore, the energy feed rates are 1 GW for wood and 40 GW for oil (assuming a higher heating value of 17.5 GJ/tonne for wood and 6.1 GJ per barrel of oil equivalent (BOE)). The scale difference is caused by the high transport cost of biomass ($0.10/km/tonne)3 combined with the slow natural growth of lignocellulosics which limits the harvest radius around a production facility. The second techno-economic barrier is the high operating cost to convert lignocellulosics into energy dense biofuels. This is caused by the natural resistance of woody biomass to decomposition, i.e. the “recalcitrance” to convert lignocellulosics into monosugars and lignin at high yield4 and/or by the high cost to remove oxygen from lignocellulosics.5
From the perspective of an overall material balance, there are two practical forms in which oxygen can be removed: either as CO2 or as H2O.6 The advantage of removing oxygen from biomass with a minimal amount of hydrogen may be quantified by considering the following general reaction for deoxygenating cellulose to an aliphatic hydrocarbon (H/C ratio = 2/1):
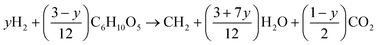 | (1) |
 | ||
| Fig. 1 Effects of hydrogen usage for deoxygenation of cellulose on carbon efficiency, mass efficiency, and energy efficiency. | ||
Important observations from Fig. 1 are that it is more energy efficient to remove oxygen as CO2 than H2O. The energy content of the products compared to the original energy content of the feedstock decreases from a maximum of 90% to 83% as hydrogen is used to remove the oxygen. Also the maximum energy efficiency is accompanied by only 35% mass efficiency and 67% carbon efficiency. This is to be expected since the goal is to increase the energy density from the feed of 17.5 MJ kg−1 to the product of 45 MJ kg−1 (diesel). Thus this simple analysis shows that it is advantageous to remove oxygen as CO2 and minimize the use of hydrogen for deoxygenation if possible. Without using hydrogen for deoxygenation, the theoretical hydrocarbon yield is approximately 2.6 BOE/MT cellulose.
A recent estimate of the capital cost of present cellulosic biofuel demonstration and commercial projects in the US is about 5 times higher than the $2.0 to $3.0 per annual gallon for corn ethanol.7 Thus, for cost-competitive production of lignocellulosic biofuels it is necessary to develop efficient, small scale conversion processes which can be located close to the biomass source.8
There are a number of biological and thermochemical biomass conversion technologies which can produce liquid fuels such as methanol, dimethyl ether, biodiesel, ethanol, butanol and pyrolysis.9–12 Some of these products can also be catalytically upgraded to deoxygenated liquid hydrocarbon fuels (LHFs). In a recent review, two routes were identified with potential to achieve economic production of liquid hydrocarbon fuels (LHF) at high yield and low complexity on an optimum scale for lignocellulosics; whole biomass pyrolysis to bio-oil and pretreatment-hydrolysis to sugars, coupled in both cases with upgrading processes.13 The latter should be carried out with a small number of reactors and with minimum utilization of external fossil-fuel hydrogen.
In the present study a novel route to LHFs has been discovered by combining hydrolysis and pyrolysis. First levulinic acid and formic acid were produced from biomass by hydrolysis through the Biofine process,14 and then the neutralized organic salt mixture was pyrolyzed to a LHF. The uniqueness of the second step is that ketonic decarboxylation15 and deoxyhydrogenation is achieved in one step at atmospheric pressure without a catalyst or externally supplied hydrogen, while the char producing reactions which hamper upgrading of pyrolysis oil16 are minimized by the stability of the organic salts in the temperature region below the pyrolysis temperature. The current method differs from ketonization such as that which is employed in the MixAlco™ process17 because our objective is to optimize hydrocarbon production during the carboxylate salt decomposition rather than to optimize ketone production.
Ketonization of organic acid salts is one pathway in which a significant amount of oxygen can be removed as CO2. In the general ketonization reaction equation:
| R1COOH + R2COOH + M(OH)2 → R1COR2 + 2H2O + MCO3 | (2) |
Formic acid has long been known as a potential source of hydrogen for transfer hydrogenation.21 Kleinert and Barth showed that high-pressure solvolysis/pyrolysis of lignin with formic acid significantly decreases the oxygen to carbon ratio of the product and produces products with a completely different structure than the starting material.22,23 Heeres et al.24 used formic acid for transfer hydrogenation of C6 sugars to γ-valerolactone with Ru/C and trifluoroacetic acid as catalysts. Haan et al.25 reported the catalytic conversion of levulinic acid with coproduct formic acid from cellulosic biomass to produce γ-valerolactone, and more recently Kopetzki and Antonietti26 demonstrated a similar conversion to γ-valerolactone via transfer hydrogenation under hydrothermal conditions.
TDO of calcium levulinate and formate mixtures
The quality and yield of the levulinic acid TDO products can be greatly improved by adding formic acid. When formic acid is present as a reactant, a highly deoxygenated hydrocarbon oil is formed. This oil has a mass density which is lower than water and is easily phase-separated as shown in Fig. 2. The organic phase also has a neutral pH (TAN ≅ 1 mg KOH/g), viscosity of 20 cP at 23 °C and an energy density higher than 40 MJ kg−1 which is about 15% greater than that for the product of pure levulinic acid TDO. | ||
| Fig. 2 Thermal deoxygenation of a mixture of calcium formate and calcium levulinate produces a hydrocarbon oil which phase separates from water. | ||
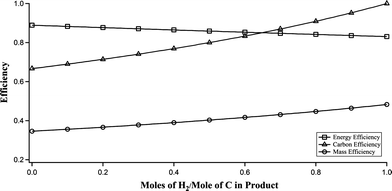
Fig. 3 presents product distributions for a range of formic/levulinic acid molar ratios. The yield of hydrocarbon oil increases and the quantity of carbon in the char decreases as the formic acid concentration increases suggesting that more hydrogen is available for deoxygenation during TDO. Furthermore, although the yield of hydrocarbon oil is about the same for formic/levulinic acid ratios of 1 and 1.5, the ratio of hydrocarbon produced to carbon fed is maximum for a ratio of 1 because carbon from the formate did not contribute to the liquid product above this point.
 | ||
| Fig. 3 Effect of formic/levulinic acid ratio on the distribution of carbon in solid char, aqueous liquid, organic liquid and gas phases. The lower error bar represents the standard deviation of solid mass remaining in the reactor. The upper error bar represents the standard deviation of the mass of hydrocarbon oil. Data for formic/levulinic acid ratio = 0 from Schwartz et al.20 | ||
The hydrocarbon oil formed at each formic acid concentration was analyzed using H NMR, 13C NMR, GC-MS, bomb calorimetry and combustion analysis. The results of these analyses are presented in Table 1 to compare the effect of formic acid concentration on the quality of the oil product. The hydrogen to carbon ratio and higher heating value do not change significantly between the four formic ratios although they may increase slightly as the formic/levulinic acid ratio increases.
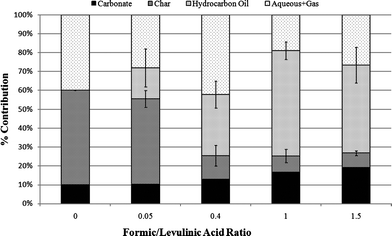
There is almost no evidence of oxygenates in the hydrocarbon oil based on 13C NMR as shown in Fig. 4 and also confirmed by H NMR. By integrating the regions in 13C NMR spectra for different acid ratios, we also determined that as formic acid concentration is increased, the oils have more conjugated double bond character, which could be either aromatics or olefins. The GC-MS data indicate that the oils contain hundreds of compounds including a significant number of alkylated aromatics and conjugated alkenes, which are consistent with the NMR analysis. The aqueous phase was found to contain hydrocarbon products, such as methyl-cyclopentenones, also found in TDO of pure levulinic acid. The non-condensable gases included CO2, CO, and C1–C3 hydrocarbons.
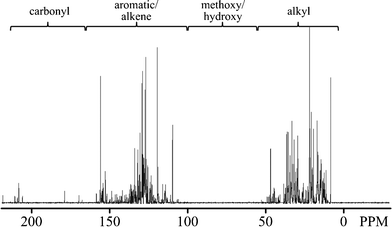 | ||
| Fig. 4 13C NMR spectra of the TDO hydrocarbon product with regions as identified in Joseph, et al.27 | ||
Increasing the quantity of formic acid as a calcium formate salt improves the yield and deoxygenation of levulinic acid TDO products. The thermogravimetric analysis shown in Fig. 5 compares the decomposition rates of calcium formate, calcium levulinate and an equimolar mixture of the two salts. In all three cases, mass loss occurs predominantly within two temperature ranges: 1) a low temperature range from 400 °C to 500 °C and 2) a high temperature range from 600 °C to 800 °C. The mass losses at low temperature correspond to decomposition of the salts, and the high temperature mass loss corresponds to decomposition of CaCO3.28 Note that the low temperature decomposition rates for all three cases overlap. Also, note that the high temperature peak is very broad for calcium levulinate, probably because of the relatively large quantity of carbonaceous residue.
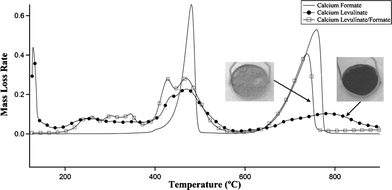 | ||
| Fig. 5 Thermogravimetric analysis of pure calcium formate, pure calcium levulinate and an equimolar mixture of formate and levulinate using a ramp rate of 10 °C min−1. The photographs of the TGA pans show that less carbon remains in the char from the formate/levulinate mixture. | ||
There are two hypotheses which might account for the increased yield of oil when decomposing formate/levulinate mixtures. First, the formate might participate with levulinate in the initial “ketonization” mechanism to form an aldehyde intermediate thus changing the subsequent vapor phase reaction pathways. A second hypothesis is that hydrogen production in this molecularly-mixed system contributes to in situ hydrodeoxygenation. It is known that calcium formate thermally decomposes to a mixture of H2, CO, and CaCO3.29 Also, it is clear in Fig. 5 that the thermal decomposition temperatures of both salts significantly overlap. Therefore, this novel technology could be applied in a broad number of applications where deoxygenation is desired.
There has been debate over the use of formic acid as a hydrogen source because of its value as an industrial chemical. In this case, however, formic acid is a co-product of cellulose hydrolysis:
 | (3) |
Further, separation and purification of the two acids would require extraction and multiple distillations, so use of the formic acid as a reagent without purification could potentially overcome the lost opportunity cost. To demonstrate this potential, TDO oils were made from unrefined levulinic acid feedstocks which were themselves produced by hydrolysis and dehydration of municipal solid wastes. These experiments demonstrated similar yields with no obvious negative effects of contaminants in the feedstock.
The current results demonstrate production of a hydrocarbon oil via a thermal pathway without the addition of hydrogen. However, the H/C ratio of this oil is only 1.3/1. It would therefore require 0.35 moles of hydrogen per mole of carbon in the product to increase the H/C ratio to 2/1 for comparison in Fig. 1.
Although this paper focuses on the use of calcium as a cation, we have used other alkali and alkali earth metals for TDO yielding similar results. The choice of metal ion might not significantly affect the yield and composition of oil, but it may affect the economics and energy requirement for the process. For instance, magnesium salts decompose at lower temperatures, which decreases the energy consumption for heating. Monovalent cations such as sodium were also employed successfully, providing evidence that both di- and mono-valent cations are useful for this method. Therefore, there may be opportunities to integrate TDO technology with existing manufacturing facilities, such as pulp mills, in which cation recovery cycles already exist.
Conclusion
Conversion of cellulosic feedstocks to hydrocarbon transportation fuels could be an important contribution to supplying future energy needs. The levulinic acid pathway, consisting of acid hydrolysis, neutralization, and TDO, is a relatively simple way which doesn't rely on heterogeneous catalysts and complex purification of feedstocks. Potential cellulosic feedstocks include forest residues, waste paper, municipal solid waste, etc. as well as traditional lignocellulosic sources such as wood and energy crops. Conversion of the five-carbon sugars to levulinic acid is also technologically feasible,30 so there is also the potential to utilize all of the carbohydrates in feedstocks if favorable economics can be demonstrated.We have improved levulinic acid TDO by adding formic acid, resulting in the production of a hydrocarbon oil in high yields. Utilizing existing hydrocracking technology, the refined TDO oil could be a direct substitute for fossil fuels such as gasoline, diesel or jet fuel. Even without further upgrading, the oil is a complex mixture of hundreds of highly deoxygenated chemical compounds with a higher heating value in excess of 40 MJ kg−1. We have investigated the effects of changing the formic/levulinic acid ratio and how that ratio affected the yield, composition and quality of the oil produced. It seems likely that a 1/1 mole ratio, which provides the highest oil yield, would be both convenient and practical from a process standpoint. However, economics based on formic acid, levulinic acid, and furfural as a valuable coproducts of hydrocarbon oil production, might favor a lower ratio.
The TDO method has many of the same advantages of fast pyrolysis; the reaction occurs at atmospheric pressure and moderately high temperature in a simple reactor. However, the product has several traits that make it superior to typical pyrolysis oils. TDO oil is produced in high yield and can be easily separated from other products such as water and char. In addition, the oil has a neutral pH, high energy density, low viscosity and a hydrogen to carbon ratio of ∼1.3. The extent of deoxygenation during TDO decreases the downstream hydrotreating required to produce a drop-in transportation fuel. Furthermore, this process does not use a catalyst, making it tolerant to trace impurities that could poison or foul precious metal catalysts.
Experimental
Reactions were conducted by forming the acid salts in a stirred 300 mL Parr reactor along with stainless steel ball bearings to aid in heat transfer and material mixing. Except for the experiment using crude levulinic acid from the Biofine pilot plant in Gorham, ME, the acid salts were prepared by mixing levulinic acid (>99%), and formic acid (>95%) with 20% excess Ca(OH)2 (>98%) in the reactor. Nitrogen was continually fed to the reactor at a rate of 100 sccm as a sweep gas, and the products were continuously condensed in a water cooled condenser (10 °C), while non-condensable products were collected in bags. The reactor was heated at approximately 10 °C min−1 while stirring at 50 rpm. Water evolved between 100–200 °C as the salts dried. Vapors evolved predominately between 375 °C and 450 °C. The temperature was then maintained constant at 450 °C until no additional product evolution was observed. Combustion analysis for hydrogen and carbon content was performed by Galbraith Laboratories, Inc. Viscosity measurements were conducted using a Brookfield viscometer. Thermogravimetry used a TA Instruments Q500 TGA. 13C NMR spectra were recorded at 100 MHz on a Varian Unity 9.4 T instrument at 29 °C. A Shimadzu GCMS-2010 with a MS-5 column was used for liquid product identification. An SRI 8610C portable GC was used to identify non-condensable products.Acknowledgements
The authors acknowledge Thomas Schwartz, Jincy Joseph, Scott Eaton, Saikrishna Mukkamala, Cody Newman, John Hessler, Nick Hill, Sedat Beis and Rachel Austin (Bates College Dept. of Chemistry) for their assistance with experiments and analyses. We also thank Steve Fitzpatrick for providing the crude levulinic acid which precipitated the current discovery, as well as Peter van Walsum, Brian Frederick, William DeSisto, and Hemant Pendse for many useful discussions. Funding for this work was provided by U.S. Dept. of Energy grant # DE-FG02-07ER46373.Notes and references
- http://forestindustries.eu/content/upms-fray-bentos-pulp-mill-uruguay, accessed October 2011.
- J. H. Gary, G. E. Handwerk and Mark J. Kaiser, Petroleum Refining Technology and Economics, 5th ed., 2007, CRC Press, Boca Raton, FL, p 10 Search PubMed.
- D. Roberts, 2008. BioRefine: Annual Conference on New Biomass Products, TEKES, September 2008, Lahti, Finland.
- P. Kumar, D. M. Barrett, M. J. Delwiche and P. Stroeve, Ind. Eng. Chem. Res., 2009, 48, 3713–3729 CrossRef CAS.
- M. M. Wright, D. E. Daugaard, J. A. Satrio and R. C. Brown, Fuel, 2009, 89, 2–10 CrossRef.
- L. Petrus and M. A. Noordermeer, Green Chem., 2006, 8, 861–867 RSC.
- B. A. Thorp, H Seamans and M. Akhtar, Major Cellulosic Biofuels/Biochemical Activities in the U.S., Proceedings of the Third Nordic Wood Biorefinery Conference NWBC 2011, Stockholm, March 22–24, p16–26.
- G.W. Huber, Chairman US National Science Foundation Workshop, 2008. Breaking the Chemical and Engineering Barriers to Lignocellulosic Biofuels: Next Generation Hydrocarbon Biorefineries, http://www.ecs.umass.edu/biofuels/Images/Roadmap2-08.pdf, accessed July 2011.
- M. Crocker and R. Andrews, The Rationale for Biofuels, Thermochemical Conversion of Biomass to Liquid Fuels and Chemicals, 2010, ed. M. Crocker, Royal Society of Chemistry, 1–25 Search PubMed.
- R. C. Brown, Introduction to Thermochemical Processing of Biomass into Fuels, Chemicals and Power, Thermochemical Processing of Biomass: Conversion into Fuels, Chemicals and Power, ed. R. C. Brown, 2011, Wiley, 1–12 Search PubMed.
- G. Olah, A. Goeppert and G. K. S. Prakash, Beyond Oil and Gas: The Methanol Economy, 2006, Wiley-VCH, Weinheim Search PubMed.
- E. Dinjus, U. Arnold, N. Dahmen, R. Höfer and W. Wach, Green Fuels-Sustainable Solutions for Transportation, Sustainable Solutions for Modern Economies, 2009, R. Höfer, ed., Royal Society of Chemistry, 125–166 Search PubMed.
- J. C. Serrano-Ruiz and J. A. Dumesic, Energy Environ. Sci., 2011, 4, 83–99 CAS.
- D. J. Hayes, S. Fitzpatrick, M. H. B. Hayes and J. R. H. Ross, Biofine process – production of levulinic acid, furfural, and formic acid from lignocellulosic feedstocks, Biorefineries Industrial Processes and Products, 2006, ed. B. Kamm, P. R. Gruber and M. Kamm, 1, 139–164 Search PubMed.
- M. Renz, Eur. J. Org. Chem., 2005, 6, 979–988 CrossRef.
- R. H. Venderbosch, A. R. Ardiyanti, J. Wildschut, A. Oasmaa and H. J. Heeres, J. Chem. Technol. Biotechnol., 2011, 85, 674–686 CrossRef.
- M. P. Landoll and M. T. Holtzapple, Biomass Bioenergy, 2010 Search PubMed.
- S. Fitzpatrick, ACS Symp. Ser., 2006, 921, 271–287 CrossRef CAS.
- B. Girisuta, L. Janssen and H. Heeres, Chem. Eng. Res. Des., 2006, 84, 339–49 CrossRef CAS.
- T. J. Schwartz, A. R. P. van Heiningen and M. C. Wheeler, Green Chem., 2010, 12, 1353–1356 RSC.
- G. Brieger and T. J. Nestrick, Chem. Rev., 1974, 74, 567–580 CrossRef CAS.
- M. Kleinert and T. Barth, Energy Fuels, 2008, 22, 1371–1379 CrossRef CAS.
- M. Kleinert, J. R. Gasson, I. Eide, A.-M. Hilmen and T. Barth, Cellulose Chem. Technol., 2011, 45(1–2), 3–12 CAS.
- H. Heeres, R. Handana, D. Chunai, C. B. Rasrendra, B. Girisuta and H. J. Heeres, Green Chem., 2009, 11, 1247–1255 RSC.
- R. H. Haan, J.-P. Lange, L. Petrus and C. J. M. Hoogenbosch, 2007. A hydrogenation process for the conversion of carboxylic acid or anester having a carbonyl group, U.S. Pat. Appl. Publ., US 20070208183 A1 20070906.
- D. Kopetzki and M. Antonietti, Green Chem., 2010, 12, 656–660 RSC.
- A. R. West, Solid State Chemistry and Its Applications, 1987, John Wiley & Sons Ltd., Great Britain, 103–104 Search PubMed.
- J. Joseph, C. Baker, S. Mukkamala, S. H. Beis, M. C. Wheeler, W. J. DeSisto, B. L. Jensen and B. G. Frederick, Energy Fuels, 2010, 24, 5153–5162 CrossRef CAS.
-
R. Reed Jr., Novel Method for the Production of Hydrogen and Hydrogen-Carbon Monoxide Mixtures, U.S. Pat., 4
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 087373, May 2, 1978.
087373, May 2, 1978. - B. V. Timokhin, V. A. Baransky and G. D. Eliseeva, Russ. Chem. Rev., 1999, 68(1), 73–84 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |