On the mechanism of selective oxidation of 5-hydroxymethylfurfural to 2,5-furandicarboxylic acid over supported Pt and Au catalysts
Sara E.
Davis
,
Bhushan N.
Zope
and
Robert J.
Davis
*
Department of Chemical Engineering, University of Virginia, 102 Engineers Way, Charlottesville, VA 22904-4741, USA. E-mail: rjd4f@virginia.edu; Fax: +1-434-982-2658; Tel: +1-434-924-6284
First published on 31st October 2011
Abstract
The mechanism of selective oxidation of aqueous 5-hydroxymethylfurfural (HMF) at high pH was studied over supported Pt and Au catalysts. Results from labeling experiments conducted with 18O2 and H218O indicated that water was the source of oxygen atoms during the oxidation of HMF to 2-hydroxymethylfurancarboxylic acid (HFCA) and 2,5-furandicarboxylic acid (FDCA), presumably through direct participation of hydroxide in the catalytic cycle. Molecular oxygen was essential for the production of FDCA and played an indirect role during oxidation by removing electrons deposited into the supported metal particles. A reaction path for HMF oxidation to FDCA was proposed.
1. Introduction
Interest in the production of commodity chemicals from renewable carbon sources instead of from fossil resources continues to grow. Oxidation of alcohols provides one such route for transformation of biorenewable feedstocks to value-added chemicals. A potential platform alcohol derived from six-carbon sugars is 5-hydroxymethylfurfural (HMF).1,2 The selective oxidation of HMF produces 2,5-furandicarboxylic acid (FDCA), a potential replacement for the terephthalic acid monomer used in the production of polyethyleneterephthalate (PET plastic).3 Thus, a variety of catalysts have been explored in the oxidation of HMF over a broad range of conditions.4–14Water is a very low-cost, polar and environmentally-benign solvent for highly oxygenated molecules derived from biomass and catalytic oxidation in water provides a sustainable, environmentally-friendly route for conversion of biomass-derived feedstocks. Gold (Au), the noblest of metals,15 is an excellent catalyst for oxidation reactions and recently, the catalytic activity of gold in liquid water has been attributed to the metal–solvent interface.16 This work further elucidates the role of water on the reactivity of metal catalysts for oxidation reactions.
The selective oxidation of HMF with molecular oxygen in liquid water is a green alternative to oxidation in organic solvents; however, a very high pH environment is often necessary for the reaction to proceed.13 The reaction scheme for oxidation of HMF through the intermediate hydroxymethylfurancarboxylic acid (HFCA) to FDCA is seen in Fig. 1. Recent work in our laboratory has shown that supported Au is more active than supported Pt for HMF oxidation, although Au is less selective to the desirable product (FDCA) under conditions of 0.15 M HMF, 0.3 M NaOH, 690 kPa O2, and 295 K.14 For gold-catalyzed oxidation of HMF, increasing the NaOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :HMF ratio as well as the Au:HMF ratio and the dioxygen pressure increased the selectivity to FDCA.5,6,14 Thus, a fundamental understanding of the roles of added base and molecular oxygen is important to developing an environmentally-friendly method for FDCA production from HMF. Moreover, additional studies on the activation of molecular oxygen on Au nanoparticles will provide new methods to control the reactivity of oxygen in organic transformations, thereby allowing the development of sustainable alternatives to traditional processes utilizing harmful inorganic oxidants.
:HMF ratio as well as the Au:HMF ratio and the dioxygen pressure increased the selectivity to FDCA.5,6,14 Thus, a fundamental understanding of the roles of added base and molecular oxygen is important to developing an environmentally-friendly method for FDCA production from HMF. Moreover, additional studies on the activation of molecular oxygen on Au nanoparticles will provide new methods to control the reactivity of oxygen in organic transformations, thereby allowing the development of sustainable alternatives to traditional processes utilizing harmful inorganic oxidants.
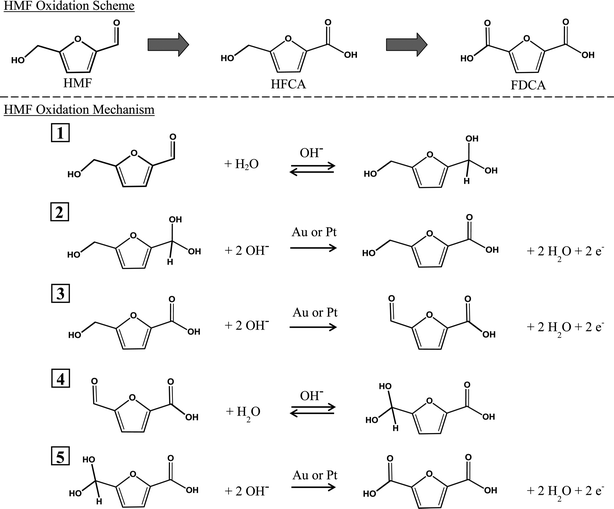 | ||
| Fig. 1 Overall reaction scheme and proposed mechanism for the oxidation of HMF in aqueous solution in the presence of excess base (OH−) and either Pt or Au. Dioxygen (not shown) serves as a scavenger of electrons that are deposited into the metal particles during the catalytic cycle. | ||
Zope et al. reported a combined experimental and computational investigation of the selective oxidation of ethanol and glycerol over Au and Pt catalysts in liquid water.16 That study provided a detailed description of the reaction path for alcohol oxidation. In addition, Casanova et al. propose a reaction scheme for the key steps in HMF oxidation.5 In the current work, we extend the studies of Zope et al.16 and Casanova et al.5 to include the oxidation of HMF over supported Au and Pt catalysts with labeled reagents 18O2 and H218O.
2. Experimental methods
2.1 Catalyst preparation
A supported gold catalyst (1.6 wt% Au/TiO2; Sample 137A) was obtained from the World Gold Council (WGC). A supported Pt catalyst (3 wt% Pt/activated carbon) was obtained from Aldrich Chemical Co. The Pt catalyst was reduced in H2 (UHP, Messer Gas) flowing at 150 cm3 min−1 for 6 h at 573 K, cooled under flowing H2 and gently exposed to air prior to use, whereas the Au catalyst was used as received. The catalysts were refrigerated and used without further pretreatment.2.2 Oxidation of HMF
The aqueous phase oxidation of HMF (Acros, ≥98% purity) was carried out in a 50 cm3 Parr Instrument Company 4592 batch reactor equipped with a glass liner. Dioxygen was either UHP (Messer Gas) or 97% 18O2 (chemical purity >99.8%, Cambridge Isotope Laboratories).In all reactions, 5.0 cm3 of the reactant solution (aqueous HMF and NaOH) were added to the reactor along with the appropriate amount of catalyst. In reactions utilizing H218O, the reactant solution was made using 97% H218O (99.8% pure, Cambridge Isotope Laboratories). The reactor was purged with flowing O2 or 18O2 and then pressurized to the desired value. A constant pressure was maintained by continuously feeding O2 or 18O2.
Because the product formation over Pt and Au as a function of reaction conditions were well described in our previous work,14 those conditions were replicated in the present study, with one exception. The dioxygen pressure in the experiments utilizing 18O2 was lower here (345 kPa) than in previous work due to the pressure limit of the 18O2 lecture bottle. Control experiments without any labeled compound were conducted prior to each experiment utilizing 18O species under conditions identical to the labeling experiments. The control experiments confirmed that the small decrease in dioxygen pressure relative to our previous study had no significant effect on product selectivity. In addition, suitable choice of NaOH:HMF ratio, O2 pressure and HMF:Au ratio resulted in a favorable product distribution to the diacid FDCA. It should be noted that although the activity and selectivity of Pt catalysts for HMF oxidation were different than those of Au catalysts, our previous work ruled out a significant influence of metal particle size and catalyst support composition.14 Therefore, we investigated different scenarios to elucidate the similarities and/or differences between the Au and Pt catalysts for oxidation. Three different scenarios were used: HMF oxidation to a majority of FDCA over Pt/C using standard conditions (0.15 M HMF, 0.3 M NaOH, 690 kPa O2, 295 K), HMF oxidation to a majority of HFCA over Au/TiO2 using standard conditions, and HMF oxidation to a majority of FDCA over Au/TiO2 in high base conditions (0.1 M HMF, 2.0 M NaOH, 2000 kPa O2, 295 K).
The samples from the oxidation reactions were filtered using 0.2 μm PTFE filters. The product analysis was conducted using a Waters e2695 high performance liquid chromatograph (HPLC) at 308 K equipped with refractive index and UV/Vis detectors. The HPLC utilized a Bio-Rad Aminex HPX-87H column and 5 mM H2SO4 flowing at 0.5 cm3 min−1 to perform the separation.
Mass spectrometry was performed using a Waters Micromass ZQ quadrupole mass spectrometer (in electronegative mode) using a direct injection of the product mixture. Prior work in our lab indicated that exchange of oxygen between the water and the product can occur during HPLC analysis.16 This exchange process was avoided by direct injection of the product into the mass spectrometer. Known compounds, in aqueous solution with NaOH, were directly infused into the mass spectrometer to obtain reference spectra. The parent peak for HFCA appeared at a mass-to-charge ratio (m/z) of 141 (mass of HFCA 142 minus 1, since H+ was removed during ionization). A sodium adduct of HFCA also appeared at m/z of 163 (mass of HFCA with one Na atom in place of one H atom, minus one proton removed during ionization). In the case of FDCA, a prominent peak appeared at m/z of 177, which corresponds to the mass of FDCA with a Na atom in place of one H atom, minus one proton removed during ionization.
To confirm that the products of HMF oxidation did not exchange O appreciably with liquid water, unlabeled HFCA and FDCA were dissolved separately in H218O in the presence of NaOH and Au/TiO2 for the duration of a reaction experiment (6 h in the case of 0.3 M NaOH and 22 h in the case of 2.0 M NaOH). Separate control experiments were conducted in both 0.3 M and 2.0 M concentrations of NaOH to replicate experimental conditions used for the reactions. The mass spectra of HFCA and FDCA in control solutions confirmed that negligible amounts of 18O from labeled water (H218O) were incorporated into the products at standard and high base conditions.
The presence of hydrogen peroxide formed in a sample reaction mixture was evaluated by a colorimetric method.17 First, a 1 cm3 sample of the filtered reaction product was immediately acidified with 1 cm3 of 0.5 M H2SO4, to which 0.1 cm3 of TiO(SO4) (15 wt% in dilute H2SO4, Aldrich) was added. Absorbance was subsequently measured at 405 nm on a Varian Cary 3E UV-vis spectrometer. A calibration curve of absorption versusH2O2 concentration was prepared by diluting a standard mixture of 30 wt% H2O2.18 The lower limit of H2O2 detection was ∼0.005 mM.
3. Results and discussion
The oxidation of HMF catalyzed by Au/TiO2 under standard conditions resulted in a majority of HFCA (≥98% selectivity) being produced after 6 h, as the oxidation of HMF to HFCA is rapid over Au.14 Previous studies showed no increase in selectivity to the diacid even if the reaction time was extended to 24 h.14 To understand the role of O2 during oxidation, an oxidation experiment over Au/TiO2 was carried out using 18O2. The major product of oxidation, HFCA, under 18O2 pressure in H216O showed no incorporation of 18O atoms (Fig. 2a). A control experiment without O2 was carried out for HMF oxidation over Au/TiO2 under standard reaction conditions. The major product, HFCA, was still obtained in significant selectivity (65%) with the rest of the product mixture being composed of 2,5-bishydroxymethylfuran (BHMF). Evidently, in the absence of O2, HMF is both oxidized (to HFCA) and reduced (to BHMF) in the basic reaction medium via the Cannizzaro reaction. Similarly, Gorbanev, et al. found 100% conversion of HMF and 51% selectivity to HFCA (selectivity to BHMF = 38%, selectivity to levulinic acid = 11%) over 18 h under similar oxygen-free conditions.6 In the presence of O2, the rate of oxidation of the aldehyde side chain of HMF on the Au catalyst was significantly greater than the Cannizzaro reaction so that nearly complete selectivity to the monoacid HFCA product was observed. To understand the role of water, HMF oxidation was carried out in labeled water, H218O. The analysis of the products from reactions under 16O2 and H218O showed peaks at m/z 145 and 167, corresponding to HFCA and the Na-adduct of HFCA with incorporation of two 18O atoms (Fig. 2a).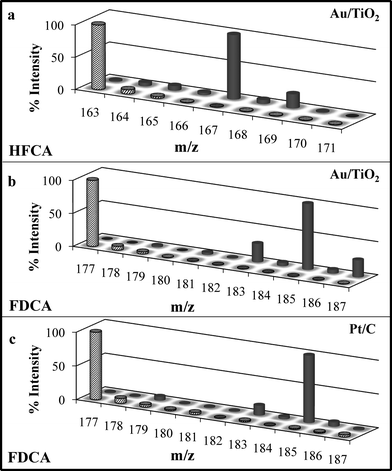 | ||
| Fig. 2
Mass spectra of major products from the oxidation of HMF (ionized in electronegative mode). Striped bars indicate experiments conducted in 18O2 and H216O; solid bars indicate experiments conducted in 16O2 and H218O. In all cases, HMF conversion = 100% and T = 295 K. m/z of major products without 18O: HFCA = 141; Na Adduct of HFCA = 163; Na Adduct of FDCA = 177. (a) Catalyst: Au/TiO2; majority product after 6 h: HFCA. Experimental conditions (both cases) HMF:Au = 150; NaOH:HMF = 2. In 16O2, P = 690 kPa; in 18O2, P = 345 kPa. (b) Catalyst: Au/TiO2; majority product after 22 h: FDCA. Experimental conditions (both cases) HMF:Au = 100; NaOH:HMF = 20. In 16O2, P = 2000 kPa; in 18O2, P = 345 kPa. (c) Catalyst: Pt/C; majority product after 6 h: FDCA. Experimental conditions (both cases) HMF:Pt = 150; NaOH:HMF = 2. In 16O2, P = 690 kPa; in 18O2, P = 345 kPa. | ||
Interestingly, incorporation of multiple 18O atoms in the acid product was observed during reaction with H218O. Because the reaction products HFCA and FDCA did not exchange O with water within the time scale of the reaction, the appearance of 18O in the products during reactions performed in H218O suggests that aqueous-phase oxidation proceeds through a geminal diol formed by the reaction of the aldehyde with the solvent. It is well known that aldehydes in water and base (OH−) rapidly undergo reversible hydration in two steps: nucleophilic addition of a hydroxide ion to the carbonyl group, followed by proton transfer from water to the alkoxide ion intermediate.19 The reversibility of geminal diol formation accounts for two 18O atoms found in the HFCA product formed during HMF oxidation (see Fig. 1, step 1). Incorporation of multiple 18O atoms in reactions performed with H218O is consistent with the findings of similar studies of ethanol and glycerol oxidation.16
To obtain high yields of the desired product (FDCA), higher NaOH concentration, catalyst loading, and dioxygen pressure were required.14 Thus, reaction conditions (0.1 M HMF, 2.0 M NaOH, 2000 kPa O2, 295 K) were utilized for the next set of experiments, except in the case of 18O2 (P = 345 kPa, due to the pressure limit of the lecture bottle). The product selectivity was not significantly affected by the lower dioxygen pressure (Table 1). The reaction time was increased to 22 h to accommodate the slow conversion of HFCA to FDCA. The major product under these conditions was FDCA (selectivity ≥69%). Again, the analysis of the product FDCA (Fig. 2b) showed incorporation of 18O into the product during the experiment with 16O2 and H218O but not with labeled gaseous oxygen, 18O2 and H216O. The FDCA peak appeared at m/z equal to 185, indicating four 18O atoms were incorporated into the Na-adduct of the product. Although the reaction conditions significantly affected the product distribution during HMF oxidation, the mode of oxygen incorporation was unchanged.
| Catalyst | 18O label | Molar ratio HMF:metal |
Molar ratio NaOH:HMF |
O2P (kPa) | Time (h) | SHFCAb (%) | SFDCAc (%) |
|---|---|---|---|---|---|---|---|
| a Conversion of HMF is 100% in all cases. b Selectivity to HFCA. c Selectivity to FDCA. | |||||||
| Au/TiO2 | none | 150 | 2 | 345 | 6 | 97 | 3 |
| Au/TiO2 | 18O2 | 150 | 2 | 345 | 6 | 98 | 2 |
| Au/TiO2 | H218O | 150 | 2 | 690 | 6 | 99 | 1 |
| Au/TiO2 | none | 100 | 20 | 345 | 22 | 35 | 65 |
| Au/TiO2 | 18O2 | 100 | 20 | 345 | 22 | 30 | 69 |
| Au/TiO2 | H218O | 100 | 20 | 2000 | 22 | 21 | 79 |
| Pt/C | none | 150 | 2 | 345 | 6 | 33 | 67 |
| Pt/C | 18O2 | 150 | 2 | 345 | 6 | 32 | 68 |
| Pt/C | H218O | 150 | 2 | 690 | 6 | 37 | 63 |
A control experiment conducted in the absence of O2 but under otherwise the same high base conditions (2.0 M NaOH) resulted in 100% conversion of HMF with 85% yield of HFCA. Moreover, simply dissolving HMF in 2.0 M NaOH solution (no O2 or metal catalyst) gave 97% conversion of HMF and 28% yield of HFCA, with the remainder of species being decomposition products such as levulinic acid and BHMF. Apparently, the presence of O2 was necessary to produce FDCA from HFCA in high base conditions over Au/TiO2 catalysts. Molecular oxygen was not needed to convert HMF to HFCA.
Platinum is known to deprotonate an alcohol to an alkoxy intermediate under neutral or mildly alkaline conditions.20 Thus, HMF oxidation was also studied over a Pt/C catalyst at standard conditions and was ≥60% selective to FDCA after 6 h (Table 1). The mass spectra corresponding to FDCA from labeling experiments with 18O are reported in Fig. 2c. Mass spectrometry analysis of the products of HMF oxidation under 18O2 pressure in H216O contained peaks at m/z values of 141, 163, and 177, corresponding to HFCA, the Na-adduct of HFCA and the Na adduct of FDCA, respectively, with no incorporation of 18O atoms. However, analysis of products from reactions under 16O2 pressure in H218O revealed peaks at 145, 167, and 185, corresponding to HFCA with the incorporation of two 18O atoms, the Na-adduct of HFCA with the incorporation of two 18O atoms, and the Na adduct of FDCA with the incorporation of four 18O atoms. Higher selectivity to the desired product, FDCA, observed here with Pt/C at standard conditions of low base and low catalyst loading confirms the noble nature of Au compared to Pt. The results also suggest that the water solvent also plays an important role during Pt-catalyzed oxidation, despite the recognized ability of Pt to dissociate O2.
A proposed mechanism for the oxidation of HMF is depicted in Fig. 1. As discussed above, molecular oxygen was not essential for oxidation of the aldehyde side-chain of HMF to produce HFCA. However, control experiments indicated that base and a metal catalyst were required to produce FDCA at the reaction temperature used here. The aldehyde side-chain is believed to undergo rapid reversible hydration to a geminal diolvia nucleophilic addition of a hydroxide ion to the carbonyl and subsequent proton transfer from water to the alkoxy ion intermediate (Fig. 1, step 1). This step accounts for the incorporation of two 18O atoms in HFCA when the reaction is performed in H218O. The second step is the dehydrogenation of the geminal diol intermediate, facilitated by the hydroxide ions adsorbed on the metal surface, to produce the carboxylic acid (Fig. 1, step 2).
An oxidation experiment performed in the absence of base over both Au and Pt catalysts resulted in no conversion of HMF, indicating the important role of OH− during oxidation at 295 K. Production of the desired product, FDCA, requires further oxidation of the alcohol side-chain of HFCA. Base is believed to deprotonate the alcohol side-chain to form an alkoxy intermediate, a step that may occur primarily in the solution.16Hydroxide ions on the catalyst surface then facilitate the activation of the C–H bond in the alcohol side-chain to form the aldehyde intermediate, 5-formyl-2-furancarboxylic acid (FCA) (Fig. 1, step 3). The next two steps (Fig. 1, steps 4 and 5) oxidize the aldehyde side-chain of FCA to form FDCA. These two steps are expected to proceed analogously to steps 1 and 2 for oxidation of HMF to HFCA. The reversible hydration of the aldehyde group in step 4 to a geminal diol accounts for two more 18O atoms incorporated in FDCA when the oxidation is performed in H218O. Thus, the sequence in Fig. 1 explains the incorporation of all 4 18O atoms in FDCA when the reaction is performed in labeled water.
Platinum is known to activate the geminal hydrogen atoms associated with alcohols.20 Also, supported Pt catalyzes alcohol oxidation in the absence of base, albeit at a very slow rate.16 These facts explain the high selectivity to FDCA during HMF oxidation over Pt compared to Au under identical conditions. Significantly increasing the concentration of hydroxide ions available, by increasing the concentration of NaOH in the reaction medium, facilitates hydrogen abstraction reactions (both C–H and O–H) on Au surfaces, therefore increasing the rate of FDCA formation over Au catalysts.
Although the role of molecular oxygen in the oxidation mechanism is not obvious, O2 is essential to produce FDCA in significant amounts during HMF oxidation over supported metal catalysts. The precise role of O2 has been debated in literature, including direct participation of atomic oxygen during dehydrogenation or oxidation steps 21 or, more recently, as an electron scavenger undergoing reduction to peroxide species and hydroxide ions.16 Our results from isotopic labeling studies indicate that molecular oxygen is not directly incorporated into the products of HMF oxidation but instead hydroxide ions from water act as a source of oxygen. Also, a test for the presence of peroxide in the product mixture of a typical reaction over Au/TiO2 under standard conditions revealed 0.3 mM of H2O2 in solution after 1 h. The presence of peroxide during oxidation reactions over Au indicates that O2 is reduced during these reactions.22Activation of O2 occurs through formation of peroxide intermediates.23 In the next step, the peroxide intermediates likely undergo further reduction to form hydroxide species.16 Therefore, O2 is suggested to undergo reduction by removing the electrons deposited into the metal particles during the adsorption and reaction of hydroxide ions, thereby completing the catalytic redox cycle. Although the reduction of O2 to hydroxide would suggest that 18O species should eventually be found in the acid products of reactions run with 18O2 and H216O after many turnovers, no 18O was observed (Fig. 2). This can be explained by the very low amount of 18O that would be found in the unlabeled water after conversion of HMF. Under our conditions and assuming 50% conversion of HMF with 100% selectivity to FDCA, 0.0023 moles of 18OH− would be produced from the oxygen reduction reactions. The amount of 16O species present initially in the H2O is 0.28 moles; thus, the molar ratio of 18O:16O species in this example is only 0.008, which is below the sensitivity of our experiment.
4. Conclusions
The oxidation of HMF to FDCA in aqueous solution at high pH is a sequential reaction in which the aldehyde side chain is first rapidly oxidized by the solvent. In a subsequent reaction, hydroxide ions from water in the presence of Au or Pt metal catalysts promote O–H and C–H bond activation of the alcohol side chain of HMF and then add directly to aldehyde intermediates to eventually form acid products. Molecular oxygen is required to scavenge the electrons deposited into the metal catalyst particles during the reaction mechanism, thus closing the catalytic cycle.Acknowledgements
This material is based upon work supported by the National Science Foundation under Grant Nos. OISE 0730277 and EEC-0813570, and by the United States Department of Energy under Grant No. DE-FG02-95ER14549. Helpful discussions with David Hibbitts and Professor Matthew Neurock are also acknowledged.References
- J. N. Chheda, G. W. Huber and J. A. Dumesic, Angew. Chem., Int. Ed., 2007, 46, 7164–7183 CrossRef CAS.
- T. Werpy and G. Petersen, Report No. NREL/TP-510-35523, 2004.
- A. Gandini, A. J. D. Silvestre, C. Pascoal Neto, A. F. Sousa and M. Gomes, J. Polym. Sci., Part A: Polym. Chem., 2008, 47, 295–298 CrossRef.
- C. Carlini, P. Patrono, A. M. R. Galletti, G. Sbrana and V. Zima, Appl. Catal., A, 2005, 289, 197–204 CrossRef CAS.
- O. Casanova, S. Iborra and A. Corma, ChemSusChem, 2009, 2, 1138–1144 CrossRef CAS.
- Y. Y. Gorbanev, S. K. Klitgaard, J. M. Woodley, C. H. Christensen and A. Riisager, ChemSusChem, 2009, 2, 672–675 CrossRef CAS.
- N. K. Gupta, S. Nishimura, A. Takagaki and K. Ebitani, Green Chem., 2011 Search PubMed.
- M. Kroger, U. Prusse and K. D. Vorlop, Top. Catal., 2000, 13, 237–242 CrossRef CAS.
- M. A. Lilga, R. T. Hallen and M. Gray, Top. Catal., 2010, 53, 1264–1269 CrossRef CAS.
- W. Partenheimer and V. Grushin, Adv. Synth. Catal., 2001, 343, 102–111 CrossRef CAS.
- E. Taarning, I. S. Nielsen, K. Egeblad, R. Madsen and C. H. Christensen, ChemSusChem, 2008, 1, 75–78, DOI:10.1002/cssc.200700033.
- P. Verdeguer, N. Merat and A. Gaset, J. Mol. Catal., 1993, 85, 327–344 CrossRef CAS.
- P. Vinke, H. E. van Dam and H. van Bekkum, in New Developments in Selective Oxidation, ed. G. Centi and F. Trifiro, Elsevier, New York, 1990, pp.147–157 Search PubMed.
- S. E. Davis, L. R. Houk, E. C. Tamargo, A. K. Datye and R. J. Davis, Catal. Today, 2011, 160, 55–60 CrossRef CAS.
- B. Hammer and J. K. Norskov, Nature, 1995, 376, 238–240 CrossRef CAS.
- B. N. Zope, D. D. Hibbitts, M. Neurock and R. J. Davis, Science, 2010, 330, 74–78 CrossRef CAS.
- C. N. Satterfield and A. H. Bonnell, Anal. Chem., 1955, 27, 1174 CrossRef CAS.
- B. N. Zope and R. J. Davis, Top. Catal., 2009, 52 CrossRef CAS.
- F. A. Carey, Organic Chemistry, New York, 5th edn, 2003, vol. 1, ch. 17, pp.712–717 Search PubMed.
- K. Heyns, C. Rudiger and H. Paulsen, Chem. Ber., 1973, 106, 623 CrossRef CAS.
- T. Mallat and A. Baiker, Chem. Rev., 2004, 104, 3037–3058 CrossRef CAS.
- W. C. Ketchie, M. Murayama and R. J. Davis, J. Catal., 2007, 250, 264–273 CrossRef CAS.
- A. A. Gewirth and M. S. Thorum, Inorg. Chem., 2010, 49, 3557–3566 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |