Synthesis and properties of trialkyl(2,3-dihydroxypropyl)phosphonium salts, a new class of hydrophilic and hydrophobic glyceryl-functionalized ILs
Fabio
Bellina
*,
Cinzia
Chiappe
* and
Marco
Lessi
Dipartimento di Chimica e Chimica Industriale, Via Risorgimento 35, 56126, Pisa, Italy. E-mail: bellina@dcci.unipi.it; cinziac@farm.unipi.it
First published on 1st November 2011
Abstract
A series of new ionic liquids (ILs) based on trialkylglycerylphosphonium cations have been prepared starting from 3-chloropropane-1,2-diol or (2,2-dimethyl-1,3-dioxolan-4-yl)methanol, two compounds readily obtainable from glycerol (a widely available and cheap waste product). All the new phosphonium salts were characterized by NMR, IR and DSC, and their efficacy as polar additives in a typical base-promoted Baylis–Hillman reaction was also evaluated.
Introduction
According to current convention, Ionic Liquids (ILs) are compounds entirely composed of ions (like inorganic salts) that have a low melting point, usually below 100 °C. This class of substances have received great attention since the discovery, by Wilkes and Zawarotko in 1992,1 of air and water stable ILs based on the imidazolium cation. Nowadays, there is no doubt that imidazole-based ILs are a focal point both for academic and industrial research and, thanks to their peculiar properties (i.e. thermal and electrochemical stability, low vapour pressure, etc.), they have been applied in different areas of chemistry, ranging from process technologies to fine chemicals' synthesis.2 Moreover, the synthetic versatility of the imidazole core allowed the preparation of more specialized ILs, the so called Task-Specific Ionic Liquids (TSILs),3 whose structures have been tailored for well-defined applications, such as CO2 capture,4 metal dissolution,5 and catalysts' stabilization.6 However, the presence of an acidic C–H at position 2 of several imidazolium salts and a recently reported thermal instability of the same imidazolium salts limit the fields of application of this interesting class of compounds.7In order to overcome these difficulties, organic cations other than imidazolium have been suggested. Among them, phosphonium salts emerged as a very promising alternative to imidazolium analogues due to a superior thermal stability and an inertness in basic reaction media.8Phosphonium based ILs have been known since the 1980s,9 but they were scarcely used until 1990, when phosphines used as precursors became available on a large scale.10 Moreover, phosphonium-based ILs are generally insoluble and less dense than water, properties that may be useful during the work-up of the reaction mixtures.
Recently, in continuation of our studies on the synthesis and applications of new ILs starting from renewable sources,11 we became interested in preparing phosphonium-based ILs containing two vicinal hydroxyl groups. In particular, in this paper we report the preparation and the characterization of a new family of n-butyl- or n-octylphosphonium salts 1 and 2, respectively, starting from commercially avaliable trialkylphosphines (3) and 1-chloro-2,3-propandiol (4) or (2,2-dimethyl-1,3-dioxolan-4-yl)methanol (5), two derivatives of glycerol (Fig. 1).12 These new ILs have also been tested as additives in a typical Baylis–Hillman reaction, due to their stability in the basic reaction conditions.
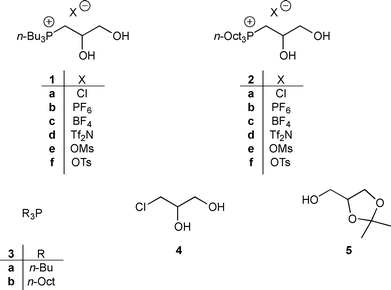 | ||
| Fig. 1 Structures of new phosphonium-based ILs 1 and 2, and of their precursors 3, 4 and 5. | ||
Experimental
NMR spectra were recorded at room temperature using a Varian 300 instrument at 300 MHz (1H), 75.7 MHz (13C), 121.4 MHz (31P) and 282.2 (19F) in DMSO-d6, CDCl3 or D2O. FT-IR spectra were recorded using a Perkin Elmer Spectrum One FTIR-ATR (400–4000 cm−1). Differential scanning calorimetry (DSC) measurements were performed on a Mettler Toledo 822e instrument according to the following procedure: the samples (5–10 mg) were sealed in aluminum pans under an inert atmosphere, and the temperature was calibrated using indium and tin as standards; the samples were then heated from 30 °C to 100 °C, at 20 °C min−1 (1st heating), cooled to the set low temperature (−50 °C) at the same scan rate (1st cooling), and heated again to the set high temperature, 200 °C at 20 °C min−1 (2nd heating). Crystallization and melting enthalpies were evaluated from the integrated areas of melting peaks. The MS and MS-MS analyses were carried out using a Perkin Elmer/Sciex API365 instrument with Turbo Ion Spray as Ion source, in flow injection analysis positive ions. GC-analyses were carried out using an Alltech AT-35 bonded FSOT column (30 m × 0.25 mm i.d.) and an Alltech AT-1 bonded FSOT column (30 m × 0.25 mm i.d.).Tributylphosphine (3a) was distilled and stored under Ar. Compounds 7a and 7b were prepared as reported in the literature.13 All the other commercially available precursors were used as received.
Tributyl(2,3-dihydroxypropyl)phosphonium chloride (1a)
A round bottomed flask was charged with deareated 1-chloro-2,3-dihydroxypropane (4, 5 g, 45.2 mmol) and tributylphosphine (3a, 14 mL, 11.31 g, 56 mmol). The resulting reaction mixture was vigorously stirred at 120 °C under argon, and the progress of the reaction was monitored by 31P NMR. After 72 h, the reaction was cooled to room temperature, then water and petroleum ether were added. The aqueous phase was extracted three times with petroleum ether, concentrated at reduced pressure and dried at 70 °C for 18 h under high vacuum to give 13.50 g (96%) of 1a as a colorless liquid.
FT-IR (neat, cm−1): ![[3 with combining low line]](https://www.rsc.org/images/entities/char_0033_0332.gif)
![[2 with combining low line]](https://www.rsc.org/images/entities/char_0032_0332.gif)
![[9 with combining low line]](https://www.rsc.org/images/entities/char_0039_0332.gif) ;2958; 2931;2872; 1464;1382;
;2958; 2931;2872; 1464;1382; ![[1 with combining low line]](https://www.rsc.org/images/entities/char_0031_0332.gif)
![[0 with combining low line]](https://www.rsc.org/images/entities/char_0030_0332.gif) ;
; ![[4 with combining low line]](https://www.rsc.org/images/entities/char_0034_0332.gif)
![[6 with combining low line]](https://www.rsc.org/images/entities/char_0036_0332.gif) ; 1H-NMR (300 MHz; DMSO; TMS):5.97 (s, 1H,O
; 1H-NMR (300 MHz; DMSO; TMS):5.97 (s, 1H,O![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) ), 5.24 (s, 1H,O), 3.90 (m, 1H, HOC), 3.38 (m, 2H,HOC2), 2.35 (m, 8H, P+
), 5.24 (s, 1H,O), 3.90 (m, 1H, HOC), 3.38 (m, 2H,HOC2), 2.35 (m, 8H, P+![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) 2), 1.46 (m, 12H, 2CH3), 0.92 (t, J = 7.0 Hz, 9H, CH3).; 13C-NMR (75.4 MHz; DMSO; TMS): 66.6 (m, HOH, H2OH); 23.8 (d, J = 16 Hz; P+CH2H2CH2CH3); 23.4 (d, J = 48 Hz, P+H2CH(OH)CH2OH); 23.2 (d, J = 4 Hz; P+CH2CH2H2CH3); 18.9 (d, J = 48 Hz, P+H2CH2CH2CH3); 13,7; 31P-NMR (121.4 MHz, DMSO; H3PO4(aq, ext)): 34.2. C15H34ClO2P (312.86); calcd. C 57.59, H 10.95; found C 57.74, H 11.00.
2), 1.46 (m, 12H, 2CH3), 0.92 (t, J = 7.0 Hz, 9H, CH3).; 13C-NMR (75.4 MHz; DMSO; TMS): 66.6 (m, HOH, H2OH); 23.8 (d, J = 16 Hz; P+CH2H2CH2CH3); 23.4 (d, J = 48 Hz, P+H2CH(OH)CH2OH); 23.2 (d, J = 4 Hz; P+CH2CH2H2CH3); 18.9 (d, J = 48 Hz, P+H2CH2CH2CH3); 13,7; 31P-NMR (121.4 MHz, DMSO; H3PO4(aq, ext)): 34.2. C15H34ClO2P (312.86); calcd. C 57.59, H 10.95; found C 57.74, H 11.00.
Tributyl(2,3-dihydroxypropyl)phosphonium hexafluorophosphate (1b)
A round bottomed flask was charged with 1a (7.00 g, 22.4 mmol) and H2O (15.0 mL). The resulting suspension was gently warmed until a solution was obtained; then, KPF6 (4.63 g, 25.1 mmol) was added in one portion. The reaction mixture was warmed at 45 °C for 18 h, then cooled to room temperature and diluted with CH2Cl2 and water. The two phases were separated, and the organic phase was washed with water to negative AgNO3 test, concentrated at reduced pressure and dried at 60 °C for 18 h under high vacuum to give 7.71 g (78%) of 1b as a colorless liquid.
FT-IR (neat, cm−1): ![[5 with combining low line]](https://www.rsc.org/images/entities/char_0035_0332.gif)
![[7 with combining low line]](https://www.rsc.org/images/entities/char_0037_0332.gif) ;
;![[8 with combining low line]](https://www.rsc.org/images/entities/char_0038_0332.gif) ; 2961; 2876; 1458; 1415; ; ; 824; 1H-NMR (300 MHz; DMSO; TMS): 5.47 (d, J = 5.1 Hz, 1H, CHO), 4.95 (t, J = 5.1 Hz, 1H, CH2O), 3.84 (m, 1H, COH), 3.36 (m, 2H, C2OH), 2.32 (m, 8H, P+C2), 1.51 (m, 12H, 22), 0.92 (t, J = 7.1 Hz, 9H); 13C-NMR (75.4 MHz; DMSO; TMS): 67.0 (d, J = 6 Hz, H2OH); 66.8 (d, J = 14 Hz, HOH); 24.0 (d, J = 16 Hz, P+CH2H2CH2CH3); 23.31 (d, J = 4, P+CH2CH2H2CH3); 23.27 (d, J = 50 Hz, P+H2CH(OH)CH2OH); 19.0 (d, J = 48, P+H2CH2CH2CH3); 13.5; 31P-NMR (121.4 MHz, DMSO, H3PO4(aq, ext)): 34.3; −143.2 (ept, J = 710 Hz); 19F-NMR (282.2 MHz, DMSO, C6F6): −70.2 (d, J = 711 Hz). C15H34F6O2P2 (422.37); calcd C 42.65, H 8.11; found C 42.81, H 8.08.
; 2961; 2876; 1458; 1415; ; ; 824; 1H-NMR (300 MHz; DMSO; TMS): 5.47 (d, J = 5.1 Hz, 1H, CHO), 4.95 (t, J = 5.1 Hz, 1H, CH2O), 3.84 (m, 1H, COH), 3.36 (m, 2H, C2OH), 2.32 (m, 8H, P+C2), 1.51 (m, 12H, 22), 0.92 (t, J = 7.1 Hz, 9H); 13C-NMR (75.4 MHz; DMSO; TMS): 67.0 (d, J = 6 Hz, H2OH); 66.8 (d, J = 14 Hz, HOH); 24.0 (d, J = 16 Hz, P+CH2H2CH2CH3); 23.31 (d, J = 4, P+CH2CH2H2CH3); 23.27 (d, J = 50 Hz, P+H2CH(OH)CH2OH); 19.0 (d, J = 48, P+H2CH2CH2CH3); 13.5; 31P-NMR (121.4 MHz, DMSO, H3PO4(aq, ext)): 34.3; −143.2 (ept, J = 710 Hz); 19F-NMR (282.2 MHz, DMSO, C6F6): −70.2 (d, J = 711 Hz). C15H34F6O2P2 (422.37); calcd C 42.65, H 8.11; found C 42.81, H 8.08.
Tributyl(2,3-dihydroxypropyl)phosphonium tetrafluoroborate (1c)
A round bottomed flask was charged with 1a (7.35 g, 23.5 mmol) and H2O (15.5 mL). The resulting suspension was gently warmed until a solution was obtained; then, NaBF4 (2.90 g, 26.4 mmol) was added in one portion. The reaction mixture was warmed at 40 °C for 18 h, then cooled to room temperature and diluted with CH2Cl2 and water. The two phases were separated, and the organic phase was washed with water to negative AgNO3 test, concentrated at reduced pressure and dried at 60 °C for 18 h under high vacuum to give 5.83 g (68%) of 1c as a pale orange liquid.
FT-IR (neat, cm−1): ; 2961; 2935; 2875; 1466; 1384; 1046; 1029; 1H-NMR (300 MHz; DMSO; TMS): 5.42 (d, J = 5.2 Hz, 1H, CHO), 4.92 (t, J = 5.4 Hz, 1H, CH2O), 3.83 (m, 1H, COH), 3.36 (m, 2H, C2OH), 2.26 (m, 8H, P+CH2), 1.45 (m, 12H, CH2CH2), 0.92 (t, J = 7.1 Hz, 9H, CH3); 13C-NMR (75.4 MHz; DMSO; TMS): 66.8 (m, HOH, H2OH); 24.0 (d, J = 16 Hz; P+CH2H2CH2CH3); 23.3 (m, P+CH2CH2H2CH3);23.2 (d, J = 50 Hz, P+H2CH(OH)CH2OH); 19.0 (d, J = 48, P+H2CH2CH2CH3); 13.7; 31P-NMR (121.4 MHz, DMSO, H3PO4(aq, ext)): 34.4; 19F-NMR (282.2 MHz, DMSO, C6F6): −147.9; −148.0. C15H34BF4O2P (364.21); calcd C 49.47, H 9.41; found C 49.53, H 9.44.
Tributyl(2,3-dihydroxypropyl)phosphonium bis(trifluoromethane)sulfonimide (1d)
A round bottomed flask was charged with 1a (3.86 g, 12.3 mmol) and H2O (8.5 mL). The resulting suspension was gently warmed until a solution was obtained; then Li(NTf)2 (3.90 g, 13.6 mmol) was added in one portion. The reaction mixture was warmed at 60 °C for 18 h, then cooled to room temperature and diluted with CH2Cl2 and water. The two phases were separated, and the organic phase was washed with water to negative AgNO3 test, concentrated at reduced pressure and dried at 60 °C for 18 h under high vacuum to give 6.80 g (99%) of 1d as a colorless liquid.
FT-IR (cm−1): ; 2965; 2938; 2877; 1467; 1347; 1178; 1133; ; ; 1H-NMR (300 MHz; DMSO; TMS): 5.44 (s, 1H, O), 4.91 (s, 1H, O), 3.84 (m, 1H, COH), 3.37 (m, 2H, C2OH), 2.31 (m, 8H, P+CH2), 1.46 (m, 12H, CH2CH2), 0.92 (t, J = 7.1 Hz, 9H, CH3); 13C-NMR (75.4 MHz; DMSO; TMS): 120.1 (q, J = 322 Hz; CF3S(O2)); 66.9 (d, J = 6 Hz, H2OH); 66.8 (d, J = 14 Hz, HOH); 23.4 (d, J = 16 Hz, P+CH2H2CH2CH3); 23.31 (d, J = 4, P+CH2CH2H2CH3); 23.28 (d, J = 40 Hz, P+H2CH(OH)CH2OH); 19.0 (d, J = 48, P+H2CH2CH2CH3); 13.5; 31P-NMR (121.4 MHz, DMSO, H3PO4(aq, ext)): 34.4; 19F-NMR (282.2 MHz, DMSO, C6F6): −78.8. C17H34F6NO6PS2 (557.55); calcd C 36.62, H 6.15; found C 36.79, H 6.13.
Trioctyl(2,3-dihydroxypropyl)phosphonium chloride (2a)
A round bottomed flask was charged with deareated 4 (4.00 g, 36.2 mmol) and trioctylphosphine (3b, 14.7 mL, 12.19 g, 32.9 mmol). The resulting reaction mixture was vigorously stirred at 120 °C under argon, and the progress of the reaction was monitored by 31P NMR. After 72 h the reaction was cooled to room temperature, then water and petroleum ether were added. The organic phase was then washed three times with water, concentrated at reduced pressure and dried at 70 °C for 18 h under high vacuum to give 15.00 g (95%) of 2a as a colorless liquid.
FT-IR (neat, cm−1):; 2955; 2915; 2855; 1465; 1377; 1280; ; ; 1H-NMR (300 MHz; DMSO; TMS): 6.00 (s, 1H, OH), 5.23 (s, 1H, OH), 3.90 (m, 1H, COH), 3.38 (m, 2H, 2OH), 2.41 (m, 8H, P+C2), 1.49 (m, 36H, 2-alk), 0.87 (m, 9H., CH3); 13C-NMR (75.4 MHz; DMSO; TMS): 66.6 (m, H2OH, HOH); 31.7; 30.6 (d, J = 16 Hz, P+CH2H2CH2CH2-); 28.9; 28.7; 23.5 (d, J = 50 Hz, H2CH(OH)CH2OH); 22.5; 21.2(d, J = 4.5 Hz, P+CH2CH2H2CH2–); 19.1 (d, 47 Hz, P+H2CH2CH2CH2-); 14.2; 31P δ (121.4 MHz; DMSO; H3PO4(aq, ext)): 34.2. C27H58ClO2P (481.17); calcd C 67.40, H 12.15; found C 67.62, H 12.19.
Trioctyl(2,3-dihydroxypropyl)phosphonium hexafluoroPhosphate (2b)
A round bottomed flask was charged with 2a (5.0 g, 10.4 mmol) and acetone (10.0 mL), then KPF6 (2.93 g, 16.0 mmol) was added in one portion under stirring. The resulting reaction mixture was stirred at room temperature for 24 h, filtered and concentrated at reduced pressure. The resulting crude product was diluted with CH2Cl2 to remove the excess of KPF6, filtered and concentrated at reduced pressure to give 5.82 g (92%) of 2b as a colorless solid.
Mp = 48 °C; FT-IR (neat, cm−1): ; 2926; 2856; 1464; 1408; 1378; ; ; 832; 1H δ (300 MHz; DMSO; TMS): 5.33 (s, 1H, OH), 4.77 (s, 1H, OH), 3.85 (m, 1H, OH), 3.38 (m, 2H, 2OH), 2.29 (m, 8H), 1.5 (m, 36H, 2-alk), 0.88 (t, J = 6.5 Hz, 9H); 13C-NMR (75.4 MHz; DMSO; TMS): 66.9 (d, J = 6 Hz, H2OH); 66.7 (d, J = 14 Hz, HOH); 31.9; 30.8 (d, J = 15 Hz, P+CH2H2CH2CH2-); 29.1; 28.8; 23.3 (d, J = 50 Hz, H2CH(OH)CH2OH); 22.7; 21.3 (d, J = 4 Hz, P+CH2CH2H2CH2-); 19.2 (d, J = 47 Hz, P+H2CH2CH2CH2-); 14.4; 31P-NMR (121.4 MHz, DMSO, H3PO4(aq, ext)): 35.8; −141.6 (hept, J = 709 Hz); 19F-NMR (282.2 MHz; DMSO; C6F6): −70.4 (d, J = 710 Hz). C27H58F6O2P2 (590.69); calcd C 54.90, H 9.90; found C 55.02, H 9.96.
TriOctyl(2,3-dihydroxypropyl)phosphonium tetrafluoroborate (2c)
A round bottomed flask was charged with 2a (5.00 g, 10.4 mmol) and a mixture of ethanol and water (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) (8.0 mL), then NaBF4 (1.71 g, 15.6 mmol) was added in one portion under stirring. The resulting reaction mixture was stirred at room temperature for 24 h and diluted with CH2Cl2 and water. The organic extract was repeatedly washed with water, concentrated at reduced pressure and dried at 60 °C for 18 h under high vacuum to give 4.24 g (77%) of 2c as a pale yellow liquid.
:1) (8.0 mL), then NaBF4 (1.71 g, 15.6 mmol) was added in one portion under stirring. The resulting reaction mixture was stirred at room temperature for 24 h and diluted with CH2Cl2 and water. The organic extract was repeatedly washed with water, concentrated at reduced pressure and dried at 60 °C for 18 h under high vacuum to give 4.24 g (77%) of 2c as a pale yellow liquid.
FT-IR (neat, cm−1):; 2925; 2855; 1465; 1411; 1378; 1051; 1031; 1H-MNR (300 MHz; DMSO; TMS): 5.46 (s, 1H, OH), 4.98 (s, 1H, OH), 3.84 (m, 1H, OH), 3.38 (m, 2H, 2OH), 2.33 (m, 8H, P+2), 1.48 (m, 36H, 2-alk), 0.88 (t, J = 6.5 Hz, 9H, CH3); 13C-NMR (75.4 MHz; DMSO; TMS): 66.7 (m, H2OH, HOH); 31.8; 30.7 (d, J = 15 Hz, P+CH2H2CH2CH2–); 29.0; 28.8; 23.3 (d, J = 50 Hz; H2CH(OH)CH2OH); 22.6; 21.2 (d, J = 4 Hz, P+CH2CH2H2CH2-); 19.1 (d, J = 47 Hz, P+H2CH2CH2CH2-); 14.3; 31P-NMR (121.4 MHz; DMSO; H3PO4(aq, ext)): 35.9; 19F δ (282.2 MHz; DMSO; C6F6): −148.36; −148.41. C27H58BF4O2P (532.53); calcd C 60.90, H 10.98; found C 61.02, H 11.01.
Trioctyl(2,3-dihydroxypropyl)phosphonium bis(trifluoromethane)sulfonimide (2d)
A round bottomed flask was charged with 2a (4.00 g, 8.3 mmol) and a mixture of ethanol and water (2:1) (6.0 mL) then Li(NTf)2 (2.65 g, 15.6 mmol) was added in one portion under stirring. The resulting reaction mixture was stirred at 60 °C for 18 h and diluted with CH2Cl2 and water. The organic extract was repeatedly washed with water, concentrated at reduced pressure and dried at 60 °C for 18 h under high vacuum to give 5.67 g (94%) of 2d as a colorless liquid.
FT-IR (neat, cm−1): 3509; 2927; 2858; 1465; 1409; 1348; 1179; 1134 1055; 1H-NMR (300 MHz; DMSO; TMS): 5.53 (s, 1H, OH), 5.05 (s, 1H OH), 3.83 (m, 1H, OH), 3.38 (m, 2H, 2OH), 2.36 (m, 8H, P+CH2), 1.47 (m, 36H, CH2-alk), 0.88 (t, J = 6.6 Hz, 9H, CH3); 13C-NMR (75.4 MHz; DMSO; TMS): 119.5 (q, J = 320 Hz, CF3S(O2)); 66.4 (d, J = 6 Hz, H2OH); 66.2 (d, J = 14 Hz, HOH); 31.3; 30.2 (d, J = 15 Hz, P+CH2H2CH2CH2–); 28.5; 28.3; 22.8 (d, J = 50 Hz, H2CH(OH)CH2OH); 22.1; 20.7 (d, J = 4.5 Hz, P+CH2CH2H2CH2–); 18.6 (d, J = 47 Hz, P+H2CH2CH2CH2-); 13.8; 31P-NMR (121.4 MHz; DMSO; H3PO4(aq, ext)): 34.3; 19F-NMR (282.2 MHz; DMSO; C6F6): −79.1. C29H58F6NO6PS2 (725.87); calcd C 47.99, H 8.05; found C 47.77, H 8.01.
Tributyl(2,3-dihydroxypropyl)phosphonium mesylate (1e)
A round bottomed flask was charged with deareated 7a (5.00 g, 25 mmol) and 3a (7.1 mL, 5.76 g, 27 mmol). The reaction mixture was vigorously stirred at 115 °C under argon, and the conversion of the reactants was monitored by 31P-NMR. After 72 h the reaction was cooled at room temperature, and the crude product was washed three times with petroleum ether at 50 °C, concentrated at reduced pressure and dried at 50 °C for 18 h under high vacuum to give 9.07 g (88%) of 8a as a pale yellow liquid [1H-NMR (300 MHz; DMSO; TMS): 4.38 (m, 1H), 4.17 (m, 1H), 3.63 (m, 1H), 2.6 (m, 2H), 2.24 (m, 9H), 1.43 (m, 18H), 0.92 (t, J = 7.2 Hz, 9H). 31P-NMR (121.4 MHz, DMSO, H3PO4(aq, ext)): 35.6]. A solution of compound 8a in water (90 mL) was warmed at 40 °C and Amberlyst 15 (0.47 g, 10% mol of H+) was then added. The reaction mixture was vigorously stirred at the same temperature and the evolution of the reaction was monitored by NMR. After 5 h the mixture was filtered on celite, concentrated at reduced pressure and dried at 70 °C for 18 h under high vacuum to give 8.18 g (100%) of 1e as a pale yellow liquid.
FT-IR (cm−1): ; 2959, 2933; 2873; 1465; 1417; 1214; 1170; 1094; 1036;. 1H-NMR (300 MHz; DMSO; TMS): 5.43 (s, 2H, OH), 3.87 (s, 1H, COH), 3.44 (m, 1H), 3.30 (m, 1H), 2.08 (m, 8H, P+C2), 1.44 (m, 12H, CH2CH2), 0.92 (t, J = 7.0 Hz, 9H, CH3);13C-NMR (75.4 MHz; DMSO; TMS): 66.7 (m, H2OH, HOH); 40.1; 23.9 (d, J = 16 Hz, P+CH2H2CH2CH3); 23.32 (d, J = 50 Hz, P+H2CH(OH)CH2OH); 23.30 (d, J = 4, P+CH2CH2H2CH3); 18.9 (d, J = 48, P+H2CH2CH2CH3); 13.8; 31P δ (121.4 MHz; DMSO; H3PO4(aq, ext)): 34.4. C16H37O5PS (372.50); calcd C 51.59, H 10.01; found C 51.51, H 9.97.
Tributyl(2,3-dihydroxypropyl)phosphonium tosylate (1f)
A round bottomed flask was charged with deareated 7b (5.73 g, 20 mmol) and 3a (6.0 mL, 4.86 g, 24 mmol). The reaction mixture was vigorously stirred at 115 °C under argon, and the conversion of the reactants was monitored by 31P-NMR. After 48 h the reaction was cooled at room temperature, and the crude product was treated with water and petroleum ether. The aqueous phase was concentrated at reduced pressure and dried at 60 °C for 18 h under high vacuum to give 9.42 g (94%) of 8b as a pale yellow liquid [1H-NMR (300 MHz; DMSO; TMS): 7.47 (d, J = 8.1 Hz, 2H), 7.11 (d, J = 8.1 Hz, 2H), 4.36 (m, 1H), 4.18 (m, 1H), 3.63 (m, 2H), 2.43 (m, 9H), 1.43 (m, 18H), 0.906 (t, J = 7.2, 9H); 31P δ (121.4 MHz, DMSO, H3PO4(aq, ext)): 34.09.]. A solution of compound 8b in water (65 mL) was warmed at 40 °C and 0.41 g (10% mol of H+) of Amberlyst 15 were then added. The reaction mixture was vigorously stirred at the same temperature and the evolution of the reaction was monitored by NMR. After 24 h the mixture was filtered on celite, concentrated at reduced pressure and dried at 70 °C for 18 h under high vacuum to give 8.60 g (100%) of 1f as a pale yellow liquid.
FT-IR (cm−1): 3339; 2959; 2932; 2872; 1599;1495; 1465; 1404;1383;1339;1217;1174; 1120; 1100; 1032; 1009; 1H-NMR (300 MHz; DMSO; TMS): 7.49 (d, J = 8.1 Hz, 2H, CH-Ar), 7.16 (d, J = 8.1, 2H, CH-Ar), 4.83 (s, 2H, OH), 3.83 (m, 1H, COH), 3.29 (m, 2H, COH), 2.33 (m, 8H, P+CH2), 1.49 (m, 12H, CH2CH2), 0.90 (t, J = 7.1 Hz, 9H, CH3); 13C-NMR (75.4 MHz; DMSO; TMS): 145.4; 137.7; 128.0; 125.4; 66.1 (m, H2OH, HOH); 23.3 (d, J = 16 Hz, P+CH2H2CH2CH3); 22.7 (d, J = 50 Hz, P+H2CH(OH)CH2OH); 22.6 (d, J = 4; P+CH2CH2H2CH3); 20.7; 18.3 (d, J = 47, P+H2CH2CH2CH3); 13.2; 31P δ (121.4 MHz; DMSO; H3PO4(aq, ext)): 34.4. C22H41O5PS (448.60); calcd C 58.90, H 9.21; found C 59.02, H 9.24.
Trioctyl(2,3-dihydroxypropyl)phosphonium mesylate (2e)
A round bottomed flask was charged with deareated 7a (5.10 g, 24 mmol) and 3b (14.6 mL, 10.70 g, 29 mmol). The reaction mixture was vigorously stirred at 120 °C under argon, and the conversion of the reactants was monitored by 31P-NMR. After 72 h the reaction was cooled at room temperature, diluted with petroleum ether and washed repeatedly with water. The organic phase was concentrated at reduced pressure to give 13 g (93%) of 8c as a pale yellow liquid [1H-NMR (300 MHz; DMSO; TMS): 4.36 (m, 1H); 4.16 (m, 1H); 3.64 (m, 1H); 2.59 (m, 2H); 2.25 (m, 9H); 1.38 (m, 42H); 0.87 (t, J = 7.2 Hz, 3H)].A solution of compound 8c in a mixture of ethanol and water (2:1) (100 mL) was warmed at 40 °C and 0.51 g (10% mol of H+) of Amberlyst 15 were then added. The reaction mixture was vigorously stirred at the same temperature and the evolution of the reaction was monitored by NMR. After 8 h the mixture was filtered on celite, concentrated at reduced pressure and dried at 70 °C for 18 h under high vacuum to give 12.10 g (100%) of 2e as a pale yellow liquid.
FT-IR (cm−1):; 2924; 2854; 1464; 1378; 1215; 1175; ; 1036;1H-NMR (300 MHz; DMSO; TMS): 5.24 (s, 2H), 3.87 (m, 1H), 3.42 (m, 2H), 2.51 (m, 11H), 1.43 (m, 36H), 0.87 (t, J = 7.1,9H); 13C δ (75.4 MHz; DMSO; TMS): 66.19 (m; H2OH, HOH); 31.3; 30.2 (d, J = 15 Hz, P+CH2H2CH2CH2–); 28.5; 28.3; 22.9 (d, J = 50, H2CH(OH)CH2OH); 22.1; 20.8 (d, J = 4 Hz,P+CH2CH2H2CH2–); 18.6 (d, J = 47 Hz, P+H2CH2CH2CH2–); 13.8; 31P δ (121.4 MHz; DMSO; H3PO4(aq, ext)): 34.3. C28H61O5PS (540.82); calcd C 62.18, H 11.37; found C 62.29, H 11.34.
Trioctyl(2,3-dihydroxypropyl)phosphonium tosylate (2f)
A round bottomed flask was charged with deareated 7b (4.37 g, 15 mmol) and 3b (9.0 mL, 7.48 g, 20 mmol). The reaction mixture was vigorously stirred at 120 °C under argon, and the conversion of the reactants was monitored by 31P-NMR. After 48 h the reaction was cooled at room temperature, diluted with petroleum ether and washed repeatedly with water. The organic phase was concentrated at reduced pressure to give 9.00 g (91%) of 8d as a pale yellow liquid [1H-NMR (300 MHz; DMSO; TMS): 7.50 (d, J = 7.8 Hz, 2H), 7.09 (d, J = 7.8, 2H), 4.34 (m, 1H), 4.14 (m, 1H), 3.61 (m, 1H), 2.58 (m, 2H), 2.22 (m, 9H), 1.36 (m, 42H), 0.87 (t, J = 6.9 Hz]. A solution of compound 8d in a mixture of ethanol and water (2:1) (100 mL) was warmed at 40 °C and Amberlyst 15 (0.51 g, 10% mol of H+) was then added. The reaction mixture was vigorously stirred at the same temperature and the evolution of the reaction was monitored by NMR. After 8 h the mixture was filtered on celite, concentrated at reduced pressure and dried at 70 °C for 18 h under high vacuum to give 8.39 g (100%) of 2f as a colorless solid.
Mp: 39 °C; FT-IR (cm−1): 3294; 2954; 2924; 2854; 1660; 1597; 1467; 1434; 1181; 1121; 1033: 1010; 1H δ (300 MHz; DMSO; TMS):7.50 (d, J = 8.0 Hz, 2H), 7.11 (d, J = 8.0 Hz, 2H), 4.57 (sb, 1H), 3.81 (m, 1H), 3.35 (m, 1H), 2.38 (m, 9H), 1.4 (m, 36H), 0.86 (t, J = 5.9 Hz, 9H); 13C δ (75.4 MHz; DMSO; TMS): 146.1; 138.3; 128.6; 126.2; 66.9 (m, H2OH, HOH); 32.0; 38.9 (d, J = 15 Hz, P+CH2H2CH2CH2–); 29.2; 28.9; 23.5 (d, J = 50 Hz, H2CH(OH)CH2OH); 22.8; 21.4 (2C); 19.7 (d, J = 47 Hz, P+H2CH2CH2CH2–); 14.6.31P δ (121.4 MHz, DMSO, H3PO4(aq, ext)): 34.4. C34H65O5PS (616.92); calcd C 66.19, H 10.62; found C 66.11, H 10.66.
General procedure for Baylis–Hillman reaction (Table 2)
A flask was charge with 9 (125 μL, 130.5 mg, 1.23 mmol), 10 (150 μL, 137.7 mg, 1.37 mmol), DABCO (70 mg, 0.61 mmol, 50 mol%) and the phosphonium-based ionic liquid (0.123 mmol, 10 mol%), see Table 2. The reaction mixture was stirred for 24 h at room temperature, thus a small portion of the crude reaction mixture was diluted with CDCl3 and the yield of the reaction was calculated using DABCO as an internal standard. The yields are shown in Table 2.| Phosphonium salt | OH signals | Tm(°C)/ΔH(J g−1)c | Water Solubilityd | ||
|---|---|---|---|---|---|
| Y− | 1H-NMR (δ)a | FT-IR (cm−1)b | |||
| a DMSO-d6 used as solvent. The concentration of samples was 0.5 M for tributylphosphonium derivatives, 0.7 M for trioctylphosphonium derivatives; b The analysis was recorded neat; c Conditions: see experimental; d Solubility test are carried out at RT, using 20 mg of Il and 1 mL of water: y = soluble; l = low soluble; n = not soluble. | |||||
| 1a | Cl | 5.97; 5.24 | 3229 | n.d | y |
| 1b | PF6 | 5.47; 4.92 | 3579 | 52/19.4 | l |
| 1c | BF4 | 5.42; 4.92 | 3521 | n.d. | l |
| 1d | Tf2N | 5.44; 4.91 | 3525 | n.d. | n |
| 1e | MsO | 5.43 (2H) | 3326 | n.d. | y |
| 1f | TsO | 4.83 (2H) | 3339 | n.d. | y |
| 2a | Cl | 6.00; 5.23 | 3238 | 40/18 | n |
| 2b | PF6 | 5.33; 4.77 | 3572 | 48/46.2 | n |
| 2c | BF4 | 5.46; 4.98 | 3514 | n.d. | n |
| 2d | Tf2N | 5.53; 5.05 | 3509 | n.d. | n |
| 2e | MsO | 5.24 (2H) | 3326 | n.d. | n |
| 2f | TsO | 4.57 (2H) | 3294 | 39/19.9 | n |
| Entrya | IL | Yield(%)b |
|---|---|---|
| a The reactions were run using 1.23 mmol of 9, 1.37 mmol of 10, 0.61 mol of DABCO and 0.123 mmol of a phosphonium salt at RT for 24 h. b Determined by 1H-NMR analysis using DABCO as internal standard. | ||
| 1 | — | 41 |
| 2 | 1a | 64 |
| 3 | 1b | 60 |
| 4 | 1c | 66 |
| 5 | 1d | 63 |
| 6 | 1e | 60 |
| 7 | 1f | 57 |
| 8 | 2a | 54 |
| 9 | 2b | 74 |
| 10 | 2c | 66 |
| 11 | 2d | 53 |
| 12 | 2e | 44 |
| 13 | 2f | 49 |
The crude reaction mixture of entry 9 in Table 2 was purified by flash chromatography and the characterization of the product, obtained with 75% yield as a colorless liquid, is in accordance with the literature.14
The experiments of Table 3 were carried out according to the protocol used for the reactions reported in Table 2.
| Entrya | 9 (Eq.) | 10(Eq.) | DABCO (%mol) | IL (%mol) | T/°C | Yield (%)b |
|---|---|---|---|---|---|---|
| a The reactions were run for 24 h. b Calculated by 1H-NMR, using DABCO as internal standard. | ||||||
| 1 | 1 | 1.1 | 50 | 10 | rt | 74 |
| 2 | 1 | 1.1 | 50 | 50 | rt | 51 |
| 3 | 1 | 1.1 | 100 | 10 | rt | 63 |
| 4 | 3 | 1 | 50 | 10 | rt | 79 |
| 5 | 1 | 3 | 50 | 10 | rt | 57 |
| 6 | 1 | 1.1 | 50 | 10 | 50 | 44 |
| 7 | 3 | 11 | 50 | 10 | 50 | 65 |
| 8 | 1 | 1.1 | 50 | 10 | 0 | 47 |
| 9 | 3 | 1 | 50 | 10 | 0 | 62 |
Results and Discussion
The synthesis of tributyl(2,3-dihydroxypropyl)phosphonium chloride (1a) and trioctyl(2,3-dihydroxypropyl)phosphonium chloride (2a) was performed by reacting commercially available tributylphosphine (3a) or trioctylphosphine (3b) with 3-chloropropane-1,2-diol (4), as depicted in Scheme 1. Due to the easy oxidation of trialkylphosphines in the open air,15 the preparation of chlorides 1a and 2a was carried out under an inert atmosphere (see experimental).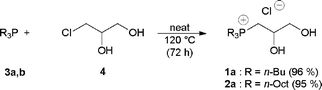 | ||
| Scheme 1 Preparation of trialkyl(2,3-dihydropropyl)phosphonium chloride 1a and 2a. | ||
Phosphonium chlorides 1a and 2a were then converted into the corresponding hexafluorophosphates 1b and 2b, tetrafluoroborates 1c and 2c, and bistriflimides 1d and 2d using metathesis reactions (Scheme 2).
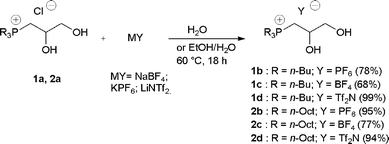 | ||
| Scheme 2 Metathesis reaction involving trialkyl(2,3-dihydropropyl)phosphonium chloride 1a and 2a. | ||
It is noteworthy to observe that the metathesis reactions involving 1a could be performed using water as the solvent, but the synthesis of trioctylphosphonium salts 2b–d required a solvent optimization due to the insolubility of 2a in H2O. Initially, the metathesis reactions involving 2a and NaBF4 or LiNTf2 were carried out in acetone, a solvent able to dissolve 2a but not the inorganic by-products (MCl).16 However, these reactions gave a mixture of products, as shown by 31P and 1H-NMR spectra of the crude reaction mixtures (Fig. 2). Besides the signals of the expected products 1c and 1d at 3.81 (CH), 3.42 and 3.29 ppm (CH2) in 1H spectrum and of a signal at 36.0 ppm in 31P-NMR (Fig. 2) groups of signals shifted downfield at 4.36 ppm (CH), 4.16 and 3.63 ppm (CH2) in 1H-NMR and a signal at 35.5 ppm in the 31P spectrum can be seen. In contrast, a single product (2b) was obtained when KPF6 was used as the reaction partner.
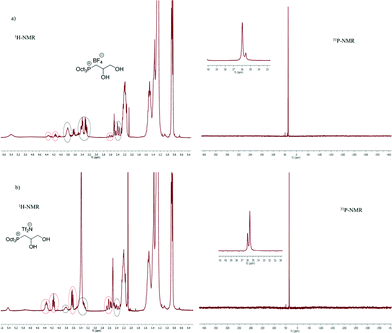 | ||
| Fig. 2 NMR spectra of the products arising from metathesis reactions of 2a in acetone. | ||
The electrospray mass (ESI-MS) analysis of the crude reaction mixure of 2c and 2d showed that the co-products of the reaction were the corresponding ketals 6a and 6b (Fig. 3), respectively arising from the reactions, probably catalyzed by Li+ or Na+ ions, of 2c and 2d with the solvent.17
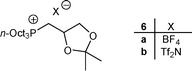 | ||
| Fig. 3 Structures of ketals 6a and 6b. | ||
However, when the metathesis reaction of 2a was carried out in a 2:1 mixture of ethanol and water the required product 2c and 2d were obtained with high chemical purity (Scheme 2).
The syntheses of mesylates 1e and 2e, and of the tosylates 1f and 2f were performed according to the reaction sequence depicted in Scheme 3, starting from commercially available 2,2-dimethyl-1,3-dioxolan-4-yl)methanol (5).
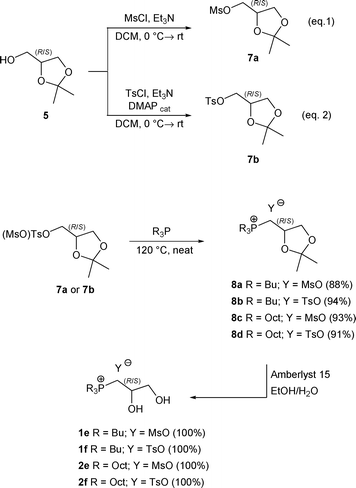 | ||
| Scheme 3 Preparation of trialkyl(2,3-dihydropropyl)phosphoniummesilates and tosylates1e–f and 2e–f, respectively. | ||
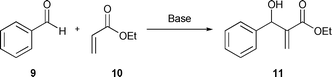 | ||
| Scheme 4 Baylis–Hillman reaction involving acetaldehyde (9) and ethylacrylate (10). | ||
In particular, the protected glycerol derivative 5 was converted into the mesylate (7a) or tosylate (7b), according to a known procedure.13 Crude 7a and 7b so obtained were then reacted with phosphines 3a or 3b, obtaining the phosphonium salts 8a–d. Finally, the required (2,3-dihydroxypropyl)phosphonium salts 1e–f and 2e–f resulted from the deprotection with Amberlyst 15® using a solvent mixture of EtOH and H2O (Scheme 3).18
It notable that phosphonium salts 8c and 8d gave 1H-NMR spectra very similar to that obtained for 6a and 6b (Fig. 3), which definitively confirmed the structures of these last compounds.
Characterization of the novel ILs
The novel phosphonium-based ILs had been fully characterized by spectroscopic analysis (NMR and IR) and by DSC analysis. Of particular interest is the analysis of the 1H-NMR and IR signals of the OH groups, which can give information on the interaction existing between the cation and the anion of any phosphonium salt. From our experimental results, summarized in Table 1, the values of the chemical shifts and of the stretching vibrations of the OH groups were influenced by the nature of the anion.In particular, the values of the OH IR stretching is between 3220 and 3340 cm−1 for anions able to form hydrogen bonds (Cl, OTs, OMs), while with poor-coordinating anions (BF4, PF6, Tf2N) the corresponding OH stretching frequencies were blue-shifted due to the absence of any interaction with these anions. A similar trend was also observed by examining the 1H-NMR spectra, where chlorides 1a and 2a displayed chemical shifts at a lower field than those observed for the corresponding phosphonium salts containing less coordinating anions (such as 1b, c and 2b, c)
Finally, DSC data showed that all the newly prepared phosphonium salts have melting points significantly lower than 100 °C, and several of them do not display any solid phase transition until temperatures as low as −50 °C
Baylis–Hillman reaction
In general, the base-promoted B–H reaction has great importance from a synthetic viewpoint, either for its high atom-economy or due to the high grade of functionalization present in the products, useful building blocks for more complex molecules.The principal drawback of the synthetic protocols involving the B–H reaction is the long reaction time generally required to drive the reaction to completion. In fact, in order to obtain good yields days or weeks are required. For this reason several studies have been carried out using small amounts of polar solvents, and in particular the use of ionic liquids evidenced a general decrease of the reaction time.19,20 This positive effect has been attributed to a stabilization of zwitterionic intermediates exerted by polar additives. The presence of H-donors, indeed, has been postulated to be beneficial for the reaction speed, as demonstrated by computational studies.21 However, it is noteworthy that the use of imidazolium-based ionic liquids as additives for the B–H reaction generally gave rise to a low isolated yield of the B–H adduct, despite the increase observed in reaction speed, probably due to the low stability of this class of ionic liquids in the reaction conditions.22
Taking into account these considerations, it seems interesting to us to test the efficiency of our new phosphonium salts as additives in a typical B–H reaction. In particular, we studied the reaction between benzaldehyde (9) and ethyl acrylate (10) in the presence of DABCO as the catalyst (Scheme 4).
The reaction was carried out using 1 equiv. of 9, 1.1 equiv. of 10, 50 mol% DABCO and 10 mol% of a phosphonium salt for 24 h (Table 2).
Data from Table 2 shows that the use of a glycol-based phosphonium salt generally leads to an increase in the yield of the B–H reaction between aldehyde 9 and acrylate 10 (compare entry 1 with entries 2–13, Table 2). In particular, when tributylphosphonium salts are used as additives, invariably higher chemical yields resulted than those observed in the absence of the ILs (entries 2–7, Table 2). In contrast, the yields of the reactions involving trioctyl phosphonium salts seem to depend on the nature of the counteranion (entries 8–13, Table 2). Specifically, high yields were obtained with octylphosphonium salts containing non-coordinating anions (PF6, BF4) (entries 9 and 10, Table 2), while when ILs containing a coordinating anion (such as Cl, OMs, or OTs) were used the yields resulting were only slightly higher than those observed when the reaction was performed in the absence of any additive (compare entries 8, 12 and 13 with entry 1, Table 2).
The best result was observed when trioctyl(2,3-dihydroxypropyl)phosphonium hexafluorophosphate 2b was used as the additive; in fact, the required B–H adduct ethyl 2-(hydroxy(phenyl)methyl)acrylate (11) was isolated in 74% yield after 24 h at room temperature.
In order to optimize the reaction conditions, we then set up a study on the influence of the reaction temperature and the molar ratio of the reagents on the efficiency of the B–H reaction, using 2b as the additive (Table 3).
From this study it emerged that the efficiency of the reaction may be improved when a molar excess of aldehyde (3 equiv.) was employed (entry 4, Table 3), while an increase in the amount of DABCO (entry 3, Table 3) or of 2b (entry 2, Table 3) led to lower yields. Similarly, a negative effect was observed when the reaction temperature was raised to 50 °C (entries 6 and 7, Table 3).
Conclusions
In conclusion, this work has demonstrated that a variety of trialkyl(2,3-dihydroxypropyl)phosphonium salts may be efficiently prepared starting from cheap and commercially available glycerol derivatives. The 2,3-dihydroxypropyl group on the phosphonium cation is able to hydrogen bond the most coordinating anions, giving salts that are generally liquid at room temperature. The interactions between ions are evidenced by NMR and IR data. The presence of long alkyl chains counterbalances the presence of the hydroxyl groups, limiting the solubility of these salts in water. All of the new compounds revealed to be efficient additives in a typical Baylis–Hillman reactions. In particular, the hydrophobic trioctyl(2,3-dihydroxypropyl)phosphonium hexafluoro-phosphate (2b) was the most efficient, giving the required Baylis–Hillman adduct in a chemical yield almost double than that obtained in the absence of any phosphonium salt.Notes and references
- J. S. Wilkes and M. J. Zaworotko, J. Chem. Soc., Chem. Commun., 1992,(13), 965 RSC.
- (a) Ionic Liquids IIIB: Fundamentals, Progress, Challenges, and Opportunities – Transformations and Processes, ed. R. D. Rogers and K. R. Seddon, ACS Symp. Ser., vol. 902, American Chemical Society, Washington D.C., 2005 Search PubMed; (b) Ionic Liquids IIIA: Fundamentals, Progress, Challenges, and Opportunities – Properties and Structure, ed. R. D. Rogers and K. R. Seddon, ACS Symp. Ser., vol. 901, American Chemical Society, Washington D. C., 2005 Search PubMed; (c) Ionic Liquids in Synthesis, ed. P. Wasserscheid and T. Welton, Wiley-VCH, Weinheim, 2nd edn, 2007 Search PubMed; (d) G. W. Meindersma, M. Maase, A. B. De Haan, Ionic Liquids, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2007 Search PubMed.
- (a) Z. Fei, T. J. Geldbach, D. Zhao and P. J. Dyson, Chem.–Eur. J., 2006, 12, 2122 CrossRef CAS; (b) S. Lee, Chem. Commun., 2006, 1049 RSC; M. Pucheault and M. Vaultier, Top. Curr. Chem., 2009, 290, 83 CAS.
- (a) E. D. Bates, R. D. Mayton, I. Ntai and J. H. Davis, J. Am. Chem. Soc., 2002, 124, 926 CrossRef CAS; (b) K. Fukumoto, M. Yoshizawa and H. Ohno, J. Am. Chem. Soc., 2005, 127, 2398 CrossRef CAS.
- A. Taubert, Top. Curr. Chem., 2009, 290, 127 CAS.
- (a) C. Chiappe, D. Pieraccini, D Zhao, Z. Fei and P. J. Dyson, Adv. Synth. Catal., 2006, 348, 68 CrossRef CAS; (b) Z. Fei, D. Zhao, D. Pieraccini, W. H. Ang, T. J. Geldbach, R. Scopelliti, C. Chiappe and P. J. Dyson, Organometallics, 2007, 26, 1588 CrossRef CAS.
- S. Sowmiah, V. Srinivadesikan, M. C. Tseng and Y. H. Chu, Molecules, 2009, 14, 3780 Search PubMed; S. Chowdhury, R. S. Mohan and J. L. Scott, Tetrahedron, 2007, 63, 2363 CrossRef.
- (a) K. J. Fraser and D. R. MacFarlane, Aust. J. Chem., 2009, 62, 309 CrossRef CAS; Multiphase Homogeneous Catalysis, ed. B. Cornils, W. A. Herrmann, I. T. Horvath, W. Leitner, S. Mercking, H. Olivier-Bourbigou, D. Vogt, Wiley-VCH, 2005, Ch. 5, p. 542 Search PubMed; (b) C. J. Bradaric, A. Downard, C. Kennedy, A. J. Robertson and Y. Zhou, Strem Chem., 2003, 20, 2 Search PubMed; (c) T. Beyersdorff, Ionic Liquid Today, 2006, 3, III Search PubMed; (d) For some applications of phosphonium-based ionic liquids, see: http://www.cytec.com/specialty-chemicals/functionalapplications2.htm.
- (a) J. F. Knifton, R. A. Grigsby Jr and J. J. Lin, Organometallics, 1984, 3, 62 CrossRef CAS; (b) J. F. Knifton, J. Am. Chem. Soc., 1981, 103, 3959 CrossRef CAS; (c) J. F. Knifton, Application: US Pat., 1981, 79-108745, 4265828, J. F. Knifton, Application: DE Pat., 1981, 80-3034019, 3034019; (d) J. F. Knifton, J. Chem. Soc., Chem. Commun., 1981, 188 RSC.
- C. J. Bradaric, A. Downard, C. Kennedy, A. J. Robertson and Y. Zhou, Green Chem., 2003, 5, 143 RSC.
- F. Bellina, C. Chiappe, A. Bertoli, B. Melai, F. Scalesse and F. Signori, Green Chem., 2009, 11, 622 RSC.
- (a) A. Ten Kate, M. J. J. Mayer, C. J. G. Cornelis, B. Kuzmanovic and C. A. M. C. Dirix, PCT Int. Appl., 2008, WO 2008074733 A1 200880626; (b) F. Jerome, Y. Poulloux and J. Barrault, ChemSusChem., 2008, 1, 586 CrossRef CAS.
- (a) F. S. Gibson, M. S. Park and H. Rapoport, J. Org. Chem., 1994, 59, 7503 CrossRef CAS; (b) P. H. G. Zarbin, E. De Beni Arrigoni, A. Reckziegel, J. A. Moreira, P. T. Baraldi and P. C. Vieira, J. Chem. Ecol., 2003, 29, 377 CrossRef CAS.
- Y. Fort, M. C. Berthe and P. Caubere, Tetrahedron, 1992, 48, 6371 CrossRef CAS.
- The chemistry of organophosphorus compounds, ed. F. R. Hartley, John Wiley & Sons, Chichester, New York, Brisbane, Toronto, Singapore, Patai. Ser., vol. 1, 1990, p. 438 Search PubMed.
- A. J. Robertson, PCT Int. Appl., 2001, WO 01/87900 A1.
- For the use of Lewis acids for the synthesis of ketals, see: (a) T.-S. Jin, Y.-R. Ma, T.-S. Li and J.-X. Wang, J. Chem. Res. (S), 1999, 268 RSC; (b) R. P. Hanzlik and M. Leinwetter, J. Org. Chem., 1978, 43, 438 CrossRef CAS; (c) P. P. Singh, M. M. Gharia, F. Dasgupta and H. G. Srivastava, Tetrahedron Lett., 1977, 5, 439 CrossRef.
- Greene's protective groups in organic synthesis, ed. P. G. M. Wuts, T. W. Greene, John Wiley & Sons, Hoboken, New Jersey, 4th edn, 2007 Search PubMed.
- (a) D. Basavaiah, B. S. Reddy and S. S. Badsara, Chem. Rev., 2010, 110, 5447 CrossRef CAS; (b) D. Basavaiah, A. J. Rao and T. Satyanarayana, Chem. Rev., 2003, 103, 811 CrossRef CAS; (c) D. Basavaiah, K. V. Rao and R. J. Reddy, Chem. Soc. Rev., 2007, 36, 1581 RSC.
- (a) A. Kumar and S. Pawar, J. Mol. Catal. A: Chem., 2004, 211, 43 CrossRef CAS; (b) H. Gong, C-Q Cai, N-F. Yang, L.-W. Yang, J. Zhang and Q.-H. Fan, J. Mol. Catal. A: Chem., 2006, 249, 236 CrossRef CAS; (c) Y.-S. Lin, C.-Y. Lin, C.-W. Liu and T. Y. R. Tsai, Tetrahedron, 2006, 62, 872 CrossRef CAS; (d) E. J. Lenardao, J. de Oliveira Feijo, S. Thurow, G. Perin and R. G. Jacob, Tetrahedron Lett., 2009, 50, 5215 CrossRef CAS; (e) C. L. Johnson, R. E. Donkor, W. Nawaz and N. Karodia, Tetrahedron Lett., 2004, 45, 7359 CrossRef CAS.
- D. Roy and R. B. Sunoj, Chem.–Eur. J., 2008, 14, 10530 CrossRef CAS.
- V. K. Aggarwal, I. Emma and A. Mereu, Chem. Commun., 2002, 1612 RSC.
| This journal is © The Royal Society of Chemistry 2012 |