Ag/Al2O3 for glycerol hydrogenolysis to 1,2-propanediol: activity, selectivity and deactivation
Jinxia
Zhou
*a,
Jing
Zhang
a,
Xinwen
Guo
b,
Jingbo
Mao
a and
Shuguang
Zhang
*ab
aCollege of Environmental and Chemical Engineering, Dalian University, Dalian, 116622, China. E-mail: jxzhou@163.com; dluzhang@126.com; Fax: 86-411-87402449; Tel: 86-411-87403214
bState Key Laboratory of Fine Chemicals, Dalian University of Technology, Dalian, 116012, China
First published on 1st November 2011
Abstract
A series of γ-Al2O3 supported silver catalysts (Ag/Al2O3) prepared with various Ag loadings and calcination temperatures were used to convert glycerol to 1,2-propanediol. A catalyst with 2 mmol Ag per gram Al2O3 and calcined at 400–500 °C presented the highest activity (glycerol conversion 46 mol%) and selectivity (96 mol%) at 220 °C, glycerol/Ag (molar ratio) = 100/2, 1.5 MPa initial H2 pressure and 10 h. Optimal prereduction, elevated reaction temperature and hydrogen pressure promote the activity, but the selectivity deteriorates at higher reaction temperatures. Excessive water is detrimental to the performance. Catalyst deactivation was observed, mainly due to Ag sintering under reducing environment. The spent catalyst could be calcined to fully recover the activity.
1. Introduction
As the worldwide production and consumption of biodiesel grows rapidly, it has been predicted that glycerol, as the major byproduct of the biodiesel manufacturing processes, will be readily available with low cost.1,2 This has triggered intensive research aiming to convert glycerol to value-added chemicals.3–5 Among these studies, the catalytic hydrogenolysis of glycerol to propanediols, which are widely used versatile speciality chemicals, is quite attractive. However, the production of 1,3-propanediol (1,3-PD) with homogeneous or heterogeneous catalysts is very challenging. It suffers from low selectivity and so far is not competitive with the fermentation route that usually presents yield above 70%.6–8 On the contrary, the catalytic conversion of glycerol to 1,2-propanediol (1,2-PD) seems viable. Two categories of catalysts, supported noble metals and transition metal oxides, have been reported in the literature for this reaction. Table 1 summarizes a few representative ones and their catalytic performance.| CatalystRef. | Conditions | Conversion (mol%) | 1,2-PD Selectivity (mol%) |
|---|---|---|---|
| Rh/SiO29 | 120 °C, 8.0 MPa initial H2 pressure, 10 h, 20 ml of 2 wt% glycerol aqueous solution, 150 mg metal catalyst. | 19.6 | 39.8 |
| Ru/TiO210 | 180 °C, 5 MPa H2, 12 h, 5 ml of 20 wt% glycerol aqueous solution, 102 mg catalyst. | 90.1 | 20.6 |
| Ru/C+Amberlyst11 | 120 °C, 8.0 MPa H2, 10 h, 20 ml of 2 wt% glycerol aqueous solution, 150 mg Ru catalyst + 300 mg Amberlyst. | 79.3 | 74.9 |
| Ru/Cs2.5H0.5[PW12O40]14 | 180 °C, 5 bar H2, 10 h, 5 ml of 20 wt% glycerol aqueous solution, 0.2 g catalyst. | 21 | 96 |
| Ru/C + Re2(CO)1015 | 160 °C, 8 MPa H2, 8 h, 10 ml of 40 wt% glycerol aqueous solution, 50 mg supported catalyst. | 59.4 | 56.6 |
| CuO/ZnO16 | 180 °C, 80 bar H2, 90 h, 15 g glycerol in 65 ml H2O, 0.08 mol Cu (50%). | 19 | 100 |
| Cu–ZnO17 | 200 °C, 4.2 MPa H2, 12 h, 15 g glycerol in 65 ml H2O, 7.5 mmol Cu (Cu/Zn atomic ratio of 1). | 22.5 | 83.6 |
| Copper-chromite18 | 200 °C, 200 psi, 24 h, 80% glycerol solution, 5 wt% of catalyst. | 54.8 | 85.06 |
| RANEY® Ni19 | 210 °C, 10 atm H2, 20 h, 8.0 g pure glycerol, 2.0 g RANEY® Ni. | 91 | 48 |
| Cu/Al2O322 | 200 °C, 1.5 MPa H2, 10 h, 50 wt% glycerol aqueous solution, Cu/glycerol molar ratio 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100. :100. |
35 | 94 |
| CuAg/Al2O323 | 200 °C, 1.5 MPa H2, 10 h, 50 wt% glycerol aqueous solution, (Cu+Ag)/glycerol molar ratio 3:100. |
27 | 96 |
| 30% w/w Ag-OMS-224 | 200 °C, 50 atm H2, 8 h, 10 g of glycerol, 40 g of 2-propanol, 0.5 g catalyst. | 59.9 | 88.9 |
Among several supported metal catalysts (metal: Rh, Ru, Pt, Pd; support: active carbon, SiO2, Al2O3), Furikado et al.9 found that Rh/SiO2 was the most active and selective one, with a glycerol conversion of 19.6% and a propanediol selectivity of 39.8%. Feng et al.10 found that a TiO2 supported Ru catalyst exhibited a markedly high activity, 90.1% glycerol conversion, but that the 1,2-PD selectivity was only 20.6%. Considering the hypothesized two-step mechanism (dehydration and hydrogenation), many researchers have opted to use bifunctional catalysts, i.e. supported noble metal catalysts combined with various acidic materials.11–14 The combination of a Ru/C and an Amberlyst resin presented a glycerol conversion of 79.3% and a propanediol selectivity of 74.7%.11 The use of Ru/C with other acidic materials, such as niobia, 12-tungstophosphoric acid (TPA) supported on zirconia, the cesium salt of TPA and the cesium salt of TPA supported on zirconia, was studied by Balaraju et al.13 They found a linear correlation between conversion and acidity on the catalysts. Alhanash et al.14 prepared a bifunctional catalyst by loading Ru onto a heteropolyacid salt Cs2.5H0.5[PW12O40], which achieved 96% selectivity to 1,2-PD at 21% glycerol conversion. As for supported bimetallic noble metal catalysts, Ma et al.15 discovered a promoting effect of Re on the catalytic performances of Ru/Al2O3, Ru/C, and Ru/ZrO2, both on the conversion of glycerol and the selectivity to propanediols.
Most transition metal oxide catalysts used for the hydrogenolysis of glycerol contain copper. Chaminand et al.16 showed that nearly 100% selectivity to 1,2-PD was achieved on a CuO/ZnO catalyst, but the activity of the catalyst was so low that it took 90 h to reach 19% glycerol conversion. Wang and Liu17 obtained a selectivity of 83.6% to 1,2-PD at 22.5% glycerol conversion. Dasari et al.18 reported that a commercial copper chromite catalyst (pre-reduced at 300 °C) converted glycerol to 1,2-PD with a selectivity of 85.0% at 54.8% conversion at 200 °C and 1.4 MPa initial H2 pressure, after 24 h. Considering the good performance of copper chromite catalysts under relatively mild reaction conditions as well as the vulnerability of noble metal catalysts to impurities in glycerol, the process using copper chromite is considered to be the most promising one for commercialization.3,18 Another catalyst that may be used at relatively low hydrogen pressure19 or even without hydrogen20 is RANEY® Ni.
In general, the disadvantage of supported noble metal catalysts is the low selectivity towards 1,2-PD. The Cu-based catalysts exhibited superior performances in terms of the selectivity towards 1,2-PD, whereas their activities are usually low. The Cu on these catalysts is initially in an oxidic state. In order to achieve a decent activity, the catalysts often need to be reduced in situ under reaction conditions with very high H2 pressure17 or to be pre-reduced to generate Cu species that are catalytically active under mild reaction conditions.21 In our previous work, we reported that a selectivity to 1,2-PD of about 97% with a glycerol conversion of about 50% was achieved on a Cu/Al2O3 catalyst, which also needed to be pre-reduced in H2.22 We also found that the introduction of Ag to the Cu/Al2O3 catalyst could eliminate the pre-reduction step.23 The present work focuses on the investigation of the catalytic activity, selectivity and deactivation of Ag/Al2O3. A molecular sieve supported Ag catalyst was recently reported in an online paper24 and a comparison with our catalyst is presented later.
2. Experimental
2.1 Catalyst preparation
Our Ag/Al2O3 catalysts were synthesized using an incipient wetness impregnation method with aqueous solutions of AgNO3 of various concentrations and γ-Al2O3 from Shandong Filiale of China Aluminium Co., Ltd., China. After impregnation, the catalysts were dried at 110 °C for 12 h and calcined in air for 3 h at 400 °C, except in the study of calcination temperature. Unless specifically stated, these catalysts were tested or characterized directly after the calcination without additional treatment. All catalysts were in powder form with particle size less than 0.32 mm in diameter. The catalysts prepared were designated as Ag/Al2O3(X), in which X represents the amount of Ag in mmol loaded on 1 g of Al2O3.2.2 Catalyst characterization
X-Ray Diffraction (XRD) patterns of the catalysts were recorded at room temperature on an X-ray diffractometer (D/max-2400) with a graphite monochromator attachment, utilizing Ni-filtered Cu-Ka radiation (40 kV, 100 mA) with a scanning speed (2θ) of 1° per minute.Nitrogen adsorption–desorption experiments for pore size distribution, pore volume, and BET surface area measurements were conducted on an ASAP2020 instrument (Micromeritics). All samples were pretreated at 350 °C under vacuum before the measurements.
Temperature Programmed Reduction (TPR) studies of the catalysts were carried out in a 10% H2/Ar gas mixture at a flow rate of 50 ml min−1 with a temperature ramp of 10 °C min−1. Before TPR tests the catalysts were dried in argon at 300 °C for 2 h. Hydrogen consumption was monitored using a thermal conductivity detector (TCD).
Characterizations with transmission electron microscopy (TEM) were carried out on a JEOL JEM-2011TEM. To prepare samples for TEM, Ag/Al2O3 samples were ground, dispersed in ethanol, and deposited onto a copper grid.
Thermogravimetric analysis (TGA) results were acquired on a TGA/SDTA851e instrument (Mettler Toledo). Samples were heated in a flow of 5% H2/N2 gas mixture (20 ml min−1) from room temperature to 700 °C with a ramp rate of 10 °C min−1.
An X-ray fluorescence (XRF) spectrometer (Thermo Scientific ARL QUANT'X EDXRF Analyzer) was used to analyze the Ag content in the spent catalyst.
2.3 Catalytic performance test
The hydrogenolysis of glycerol was carried out in a high throughput batch reactor system consisting of ten independent stainless steel autoclaves (165 ml each) with mechanical stirring. In most cases, an aqueous solution of glycerol (50 wt% concentration) prepared with pure glycerol (>99%, China National Medicines Corporation Ltd., China) and deionized water was used as feed. In a typical run, 65 g of the glycerol solution and a specified quantity of the catalyst were loaded into the reactor. The reactor was purged five times with H2 (99.99%, Dalian F.T.Z Gredit Chemical Technology Development Co., Ltd.) and pressurized with H2 to 1.5 MPa at room temperature. With stirring at 400 RPM, the mixture of the glycerol and the catalyst was heated to 220 °C and maintained for 10 h. The stirring speed was selected to eliminate the influence of external mass transfer and to avoid creating splash inside the reactor, which would make sampling and temperature control very difficult. Hydrogen was fed on demand so as to keep the total reaction pressure at 3.6 MPa during the 10 h period. After the reaction, the gas phase products were collected in a gas bag and the liquid phase products were separated from the catalyst by filtration. These products were analyzed using a gas chromatograph (GC HP5890) equipped with a flame ionization detector. The GC column used was a PEG2W capillary column (30 m × 0.32 mm × 0.5 μm) manufactured by Dalian Institute of Chemical Physics, Chinese Academy of Sciences. Solutions of n-butanol with known amounts of internal standards were prepared and used for quantification of various glycerol-derived compounds in the products. 1,2-PD was the main product with certain amount of ethylene glycol (EG). There was no 1,3-PD detected. Other byproducts in very small amount, such as 1-propanol and methane, were also identified and are listed under “Others” in the tables below. Repeated runs showed that data variation was in the range of ±5% (relative value). The conversion of glycerol and the selectivity, which have been defined in our previous publication,23 were used to evaluate the performance of each catalyst. Usually, only a trace amount of product was detected in the gas phase. The overall carbon balance in the product was >98%.3. Results and discussion
3.1 Study of catalyst preparation
| Selectivity (mol%) | ||||||
|---|---|---|---|---|---|---|
| Catalyst |
Ag:G (mol mol−1) |
Conversion (mol%) | 1,2-PD | EG | Others | Conv./Ag (mol (mol h)−1) |
| Al2O3 | 0:100 |
0 | — | — | — | — |
| Ag/Al2O3(0.5) | 0.5:100 |
20 | 94 | 3 | 3 | 4.0 |
| Ag/Al2O3(1.0) | 1:100 |
29 | 96 | 2 | 2 | 2.9 |
| Ag/Al2O3(2.0) | 2:100 |
46 | 96 | 2 | 2 | 2.3 |
| Ag/Al2O3(3.0) | 3:100 |
45 | 97 | 2 | 1 | 1.5 |
| Ag/Al2O3(4.0) | 4:100 |
41 | 96 | 2 | 2 | 1.0 |
 | ||
| Fig. 1 XRD patterns of Ag/Al2O3 catalysts with different Ag loadings. | ||
Fig. 2 shows the N2 adsorption results of Ag/Al2O3. A steady decline of surface area and pore volume was observed with the increase of Ag loading. Therefore, the low accessibility of active Ag sites due to pore blockage at high metal loading could also contribute to the low conversion of Ag/Al2O3(4.0).
 | ||
| Fig. 2 Surface areas and pore volumes of Ag/Al2O3 catalysts with different Ag loadings. | ||
Since Ag/Al2O3(2.0) presented the best catalytic performance, it was used in the following studies of calcination temperature and reaction conditions.
 | ||
| Fig. 3 The catalytic performance of AgAl2O3(2.0) calcined at different temperatures (Ag:G (molar ratio) = 2:100). | ||
In the process of catalyst preparation, Ag existed as AgNO3 on the support before calcination. Our TG analysis (not shown here) of a AgNO3 impregnated γ-Al2O3 (110 °C dried overnight) showed that the sample's weight did not stabilize until the heating temperature reached about 400 °C. Below this temperature, we believe that AgNO3 did not decompose completely, which was the case for the samples calcined at 200 and 300 °C. The X-ray photoelectron spectroscopy (XPS) analysis in our previous work23 showed that metallic Ag and Ag2O existed on the 400 °C calcined sample. The molar ratio of Ag(0) and Ag(I) is about 1. The XRD patterns of these samples in Fig. 4 did not reveal distinctive differences except that the one of the 700 °C calcined sample presented a small peak at 2θ ≈ 38.1° (metallic Ag). The surface area and pore volume of this sample are 160 m2 g−1 and 0.25 ml g−1, respectively, while the corresponding values of the 400 °C calcined sample are 223 m2 g−1 and 0.27 ml g−1. Since the phase transition temperature for γ-Al2O3 (about 1000 °C) is much higher than 700 °C, the support we used should be stable under the calcination conditions. The loss of surface area is therefore attributed to the blockage of pores due to Ag sintering caused by the calcination at 700 °C.
 | ||
| Fig. 4 XRD patterns of Ag/Al2O3(2.0) calcinated at different temperatures. | ||
The easiness of reduction of the silver species on Ag/Al2O3 varies, as shown in Fig. 5. The TPR profile of the 200 °C calcined sample had a hydrogen consumption peak centered at 150 °C, which should be the decomposition/reduction temperature of AgNO3 in hydrogen. The sample calcined at 300 °C may still have some AgNO3, which did not decompose completely. Its TPR profile displayed a shoulder peak around 110 °C besides a major peak at about 65 °C, which was also shown in the profiles of the three samples calcined at higher temperatures. These two peaks are related to the reductions of AgNO3 and Ag2O, respectively. The TPR profile of the 700 °C calcined sample is broad. This is because the sintering indicated by the XRD characterization above resulted in relatively large Ag2O crystallites, which should be easy to reduce. This is responsible for the left side of the broadened peak. Moreover, studies conducted by Yoon and coworkers using XPS and UV-vis spectroscopy concluded that high calcination temperature was beneficial to the transformation of Ag from the ionic state to the metallic state.25 It means that there will be less ionic Ag on Ag/Al2O3 samples calcined at higher temperatures. On the other hand, we believe that a small amount of silver aluminate, which is more difficult to reduce than Ag2O due to its ionic properties, was formed on the sample and contributed to the right side of the broadened peak. According to the study using X-ray Absorption Near Edge Structure by Iglesias-Juez et al.26 and to the XRD results from Nakatsuji et al.27 and from She and Flytzani-Stephanopoulos,28 certain types of silver aluminate (AgAlO2) can be formed when alumina supported silver catalysts are calcined in the temperature range 500–800 °C.
 | ||
| Fig. 5 TPR profiles of Ag/Al2O3(2.0) calcined at different temperatures. | ||
According to the TPR results above, it is likely that all Ag species would be reduced to metallic Ag under the reaction conditions so as to catalyze the hydrogenolysis. However, there should be an induction period whose length depends on the ease of reduction and affects the glycerol conversion. The catalysts calcined between 400 and 500 °C may have the easiest reducible Ag species, the shortest induction period, and therefore presented the highest glycerol conversion.
3.2 Study of reaction conditions
| Selectivity (mol%) | ||||
|---|---|---|---|---|
| Reaction T (°C) | Conversion (mol%) | 1,2-PD | EG | Others |
|
Catalyst loading: Ag:G (mol mol−1) = 2:100. |
||||
| 180 | 17 | 91 | 4 | 5 |
| 200 | 21 | 95 | 3 | 2 |
| 220 | 46 | 96 | 2 | 2 |
| 240 | 66 | 76 | 6 | 18 |
The positive effect of increasing glycerol concentration on conversion shown in Table 4 can be looked at from several aspects. As a solvent, water has a diluting effect, which is detrimental to the conversion of the reactant, glycerol. Water is also a byproduct from the hydrogenolysis. Excessive amounts of water will tend to shift the reaction equilibrium to the reactant side. It may also damage the physical structure of the catalyst, as shown later. When the water content was below 50 wt%, it was not as influential, probably because the glycerol on the catalyst surface became concentrated enough and its content was not a rate controlling factor any more.
:G = 2:100, 56 mol% of glycerol was converted. While keeping other conditions the same but increasing the catalyst usage to Ag:G = 7:100, a glycerol conversion of about 75 mol% was achieved. A combination of long reaction time and high catalyst usage, i.e. 20 h and Ag:G = 7:100, resulted in 90 mol% glycerol conversion. The 1,2-PD selectivity remained about 95 mol% in these tests, unlike the case of using high temperature (240 °C) to achieve high conversion. However, it was noticed during the reaction time study that the glycerol conversion increased only 10 mol% when the reaction time was extended from 10 h to 20 h. This can be a sign of catalyst deactivation during prolonged use.
 | ||
| Fig. 6 The effect of reaction time: Ag/Al2O3(2.0), Ag:G = 2:100. | ||
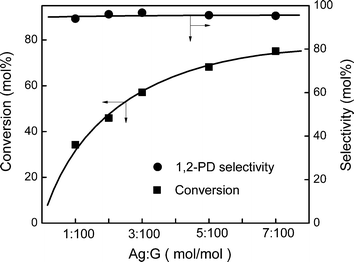 | ||
| Fig. 7 The effect of catalyst loading: Ag/Al2O3(2.0). | ||
3.3 Study of catalyst deactivation
The deactivation of Ag/Al2O3(2.0) was confirmed by the experimental results in Table 5. After a typical run at 220 °C and 1.5 MPa initial H2 pressure for 10 h, the catalyst was separated from the liquid product by filtration, washed with 100 ml deionized water and loaded into the reactor with fresh glycerol feed for another test under the same conditions. The washed catalyst (Entry 2) achieved 25 mol% conversion, about 54% of that on the fresh catalyst (Entry 1). In Entry 3, a spent catalyst was calcined at 400 °C for 3 h in air before being tested again with fresh feed. This time a glycerol conversion of 45 mol% was obtained, which can be considered to be the same as the one on the fresh catalyst considering experimental variation. The selectivity remained high on all the catalysts.| Selectivity (mol%) | ||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | Surface Area (m2 g−1) | Conversion (mol%) | 1,2-PD | EG | Others |
| 1 | Fresh | 223 | 46 | 96 | 2 | 2 |
| 2 | Spent and washed | 25 | 93 | 3 | 4 | |
| 3 | Spent, washed and calcined at 400 °C in air | 220 | 45 | 97 | 2 | 1 |
| 4 | Pretreated with H2 at 200 °C for 10 h | 52 | 96 | 2 | 2 | |
| 5 | Pretreated with H2 at 500 °C for 10 h | 45 | 97 | 2 | 1 | |
| 6 | Pretreated with H2 at 600 °C for 10 h | 35 | 97 | 2 | 1 | |
| 7 | Pretreated with H2 at 700 °C for 10 h | 15 | 96 | 2 | 2 | |
| 8 | Pretreated with N2 at 200 °C for 10 h | 44 | 96 | 2 | 2 | |
| 9 | Pretreated with 100 wt% glycerol under 1.0 MPa N2 at 200 °C for 10 h | 26 | 94 | 2 | 4 | |
| 10 | Pretreated with 50 wt% glycerol under 1.0 MPa N2 at 200 °C for 10 h | 27 | 93 | 3 | 4 | |
| 11 | Pretreated with water under 1.0 MPa N2 at 200 °C for 10 h | 56 | 7 | 89 | 5 | 6 |
There are four possible deactivation mechanisms here: support structure destruction, Ag leaching, Ag sintering, and coking. Comparison of the surface areas and pore volumes (Table 5) of the fresh catalyst and the spent and calcined one revealed that the structure was stable. The Ag content in the spent catalyst obtained from XRF analysis was 1.95 mmol g−1, very close to the value in the fresh sample. In addition, the calcination recovered essentially all the activity. All these indicate that the first two mechanisms did not play a role in the deactivation. On the other hand, TEM characterization of the fresh and the spent samples (Fig. 8(a), (b)) showed that sintering of Ag species did happen. The Ag particle size on the fresh sample is about 10 nm, while it grew to about 30 nm after use. Comparison of the XRD profiles in Fig. 9 concurs with the TEM finding. The three pronounced peaks at 2θ ≈ 38.1, 44.3, and 64.5° are the characteristic signals from large metallic Ag crystallites. The XRD characterization also showed that these signals disappeared after calcination and the XRD profile of the spent, washed and calcined sample is very similar to that of the fresh catalyst. This is because the calcination redispersed the sintered Ag and therefore rejuvenated the catalyst, as evidenced by the TEM image in Fig. 8(c). The redispersion of metals like Pt or Ni supported on γ-Al2O3, when heated in an oxidative environment in the temperature range 300–500 °C, is well known.29–33 Of course, the calcination would burn off carbonaceous species, i.e. coke, formed during the reaction. This would also regenerate the deactivated catalyst if coking was one of the sources of deactivation. The TG analysis result of the spent and washed catalyst was shown in Fig. 10. The total weight loss is about 20 wt%, among which about 8 wt% is in the range 200–600 °C. In order to eliminate the effect of water and glycerol, a similar analysis (not shown) was carried out on a Ag/Al2O3(2.0) sample that was soaked with 50 wt% glycerol solution at room temperature, washed with 100 ml water and dried at 110 °C. The weight loss of this sample between 200 and 600 °C (residual water and glycerol) is about 3 wt%. This serves as a baseline. Therefore, the net weight loss of the spent and washed sample in the temperature range is about 5 wt%. Considering the catalyst usage, we estimate that the total amount of coke formed during the reaction was no more than 0.7 wt% of the glycerol in the feed.
 | ||
| Fig. 8 TEM images of Ag/Al2O3(2.0): (a) fresh, (b) spent and washed, and (c) spent, washed and calcined at 400 °C. | ||
 | ||
| Fig. 9 XRD patterns of fresh, spent and pretreated Ag/Al2O3 catalysts. | ||
 | ||
| Fig. 10 TG analysis of the spent and washed Ag/Al2O3 catalysts. | ||
In order to pinpoint the individual contribution from the last two deactivation mechanisms, i.e.Ag sintering and coking, four more tests were carried out. In Entry 4–7 of Table 5, the fresh catalyst was reduced in hydrogen at different temperatures. The goal was to introduce sintering without coking. The XRD characterizations in Fig. 9 indicate that sintering occurred even at 200 °C and became more pronounced at elevated temperatures. It is interesting to see that the sample prereduced at 200 °C was more active than the fresh catalyst. The glycerol conversion on it was 52 mol%. The improved activity could be from the elimination of the induction (prereduction) period mentioned before in the discussion of the calcination effect. This positive effect was completely offset by the negative effect of sintering on the sample reduced at 500 °C, which showed a similar activity as the fresh catalyst (Entry 1). When the effect of sintering became dominant, the glycerol conversion dropped to 35 mol% and 15 mol% with 600 °C and 700 °C reduction, respectively.
To better quantify the correlation between deactivation and sintering, we used the intensity of the XRD signal of Ag at 2 theta 38.1° as a measurement of sintering and plotted it against the glycerol conversion on the corresponding catalysts (Fig. 11). The smooth curve indicates a clear trend of deactivation with the degree of sintering. More interestingly, the data point for the spent and washed sample fits the trend very well. This demonstrates that the deactivation under the real reaction conditions was predominantly due to Ag sintering. The 50 wt% glycerol pretreated sample is more like the spent and washed one except that it was not exposed to any hydrogen during the pretreatment. Because of the lack of the benefit from prereduction, its data point was off the curve slightly. Although the 100 wt% glycerol pretreated sample had less sintering, it was not more active than the spent and washed one or the 50 wt% glycerol pretreated one. Its data point is away from the curve. We believe that coking, which was more severe in the absence of water, contributed to the deviation in this case.
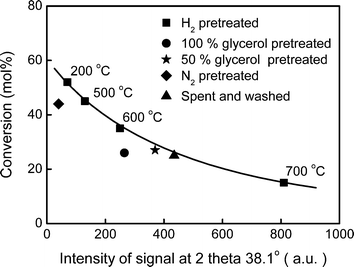 | ||
| Fig. 11 The correlation between sintering and glycerol conversion. | ||
Qu and co-workers studied the sintering of Ag on SiO2 and found that H2 treatment at low temperatures (100–300 °C) dispersed silver particles, but prolonged treatment in H2 at temperature above 300 °C induced aggregated particles.34 The temperatures observed in our study, 200 °C for slight sintering on the sample reduced in H2 and 220 °C for the pronounced one on the spent and washed catalyst, are relatively low. The difference could be from the duration of the treatment and the nature of the supports. Under the reaction conditions, the sintering is also related to the system environment. Unlike the one treated in H2, the catalyst treated in N2 at 200 °C for 10 h showed little sintering and its activity (Entry 8 in Table 5) was very similar to the fresh catalyst. Entry 9, 10 and 11 in Table 5 and the corresponding XRD patterns in Fig. 9 illustrated the effect of glycerol and water. The treatment with pure glycerol resulted in some sintering and loss of activity. Replacement of half of the glycerol with water (50 wt% glycerol solution) enhanced the sintering. The treatment with water solely (Entry 11) destroyed the catalyst structure, as indicated by the low surface area (56 m2 g−1vs. 223 m2 g−1 of the fresh one) and the activity. This result concurred with the finding from the study of the effect of glycerol concentration. On the other hand, it also manifested that 50 wt% glycerol in the system was enough to prevent the water from damaging the catalyst structure by staying in the liquid phase and formed a layer on the solid catalyst under the reaction conditions. In summary, hydrogen, glycerol, water, temperature and duration all contributed to the sintering observed here.
At the time this paper was being prepared, Yadav et al. presented a similar catalytic system in their publication.24 The Ag-OMS-2 catalysts (Ag incorporated into octahedral molecular sieve) they synthesized seem to have higher activity for glycerol hydrogenolysis to 1,2-PD. Their best catalyst has 30 wt% Ag, which was probably highly dispersed in the catalyst framework due to the co-precipitation method used. The high Ag loading and dispersion are believed to be beneficial to the activity. The stability could be good because metal sintering would be difficult if Ag is in the framework. Another factor contributing to the higher activity is the use of 2-propanol as solvent. The negative effect of water in the system has been clearly shown above. These authors observed activity loss (33–73%) too from the study of catalyst reusability in a batch reactor. The decrease of activity seems smaller in the second reuse. Their investigation using a continuous fixed bed reactor at 200 °C revealed that the glycerol conversion and 1,2-PD selectivity stabilized at about 30% and 70%, respectively, after 10 h on stream, although the corresponding numbers at 4 h were 65% and 90%, respectively. At 220 °C, the conversion (about 60%) seemed to be constant throughout the 32 h run, but the selectivity was only about 70%. We did not study either multiple reuses or continuous testing, but it is reasonable to expect that the sintering/deactivation will slow down with time while the selectivity in our work remains above 95 mol% even at 220 °C.
4. Conclusions
The preparation of Ag/Al2O3 (Ag loading, calcination temperature) and reaction conditions have been optimized for the conversion of glycerol to 1,2-PD. 2 mmol Ag per gram of Al2O3 is the desired loading and the suitable calcination is between 400 and 500 °C. About 46 mol% conversion and 96 mol% 1,2-PD selectivity were achieved at 220 °C, glycerol/Ag (molar ratio) = 100/2, 1.5 MPa initial H2 pressure and 10 h. The performance is comparable to other Cu-containing catalysts, but Ag/Al2O3 requires neither prereduction nor high hydrogen pressure. Unlike most supported noble metal catalysts, 1,2-PD selectivity on Ag/Al2O3 is much higher. Catalyst deactivation was observed and investigated. It is concluded that sintering due to the combined contribution of hydrogen, water, glycerol, temperature and reaction duration was responsible for the activity loss. The deactivation because of coking was minimal under the reaction conditions. The deactivated catalyst could be fully regenerated with a calcination in air, which redispersed the sintered Ag particles and may also burn off the coke formed.Acknowledgements
The authors are grateful to the State Key Laboratory of Fine Chemicals at Dalian University of Technology for supporting the catalyst characterization in this work under Grant KF0703. We also thank the Department of Education of Liaoning Province, China for their financial support under Grant L2010037.References
- R. G. Bray, Biodiesel Production, SRI Consulting, 2004 Search PubMed.
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044–4098 CrossRef CAS.
- M. Pagliaro, R. Ciriminna, H. Kimura, M. Rossi and C. D. Pina, Angew. Chem., Int. Ed., 2007, 46, 4434–4440 CrossRef CAS.
- A. Behr, J. Eilting, K. Irawadi, J. Leschinski and F. Lindner, Green Chem., 2008, 10, 13–30 RSC.
- C. H. Zhou, J. N. Beltramini, Y. X. Fan and G. Q. Lu, Chem. Soc. Rev., 2008, 37, 527–549 RSC.
- L. Gong, Y. Lu, Y. Ding, R. Lin, J. Li, W. Dong, T. Wang and W. Chen, Appl. Catal., A, 2010, 390, 119–126 CrossRef CAS.
- T. Kurosaka, H. Maruyama, I. Naribayashi and Y. Sasaki, Catal. Commun., 2008, 9, 1360–1363 CrossRef CAS.
- Y. Nakagawa, Y. Shinmi, S. Koso and K. Tomishige, J. Catal., 2010, 272, 191–194 CrossRef CAS.
- I. Furikado, T. Miyazawa, S. Koso, A. Shimao, K. Kunimori and K. Tomishige, Green Chem., 2007, 9, 582–588 RSC.
- J. Feng, H. Fu, J. Wang, R. Li and H. Chen, Catal. Commun., 2008, 9, 1458–1464 CrossRef CAS.
- T. Miyazawa, S. Koso, K. Kunimori and K. Tomishige, Appl. Catal., A, 2007, 318, 244–251 CrossRef CAS.
- T. Miyazawa, S. Koso, K. Kunimori and K. Tomishige, Appl. Catal., A, 2007, 329, 30–35 CrossRef CAS.
- M. Balaraju, V. Rekha, P. S. Sai Prasad, B. L. A. Prabhavathi Devi, R. B. N. Prasad and N. Lingaiah, Appl. Catal., A, 2009, 354, 82–87 CrossRef CAS.
- A. Alhanash, E. F. Kozhevnikova and I. V. Kozhevnikov, Catal. Lett., 2008, 120, 307–311 CrossRef CAS.
- L. Ma, D. He and Z. Li, Catal. Commun., 2008, 9, 2489–2495 CrossRef CAS.
- J. Chaminand, L. Djakovitch, P. Gallezot, P. Marion, C. Pinel and C. Rosier, Green Chem., 2004, 6, 359–361 RSC.
- S. Wang and H. C. Liu, Catal. Lett., 2007, 117, 62–67 CrossRef CAS.
- M. A. Dasari, P. P. Kiatsimkul, W. R. Sutterlin and G. J. Suppes, Appl. Catal., A, 2005, 281, 225–231 CrossRef CAS.
- A. Perosa and P. Tundo, Ind. Eng. Chem. Res., 2005, 44, 8535–8537 CrossRef CAS.
- A. Yin, X. Guo, W. Dai and K. Fan, Green Chem., 2009, 11, 1514–1516 RSC.
- M. Balaraju, V. Rekha, P. S. Sai Prasad, R. B. N. Prasad and N. Lingaiah, Catal. Lett., 2008, 126, 119–124 CrossRef CAS.
- L. Guo, J. Zhou, J. Mao, X. Guo and S. Zhang, Appl. Catal., A, 2009, 367, 93–98 CrossRef CAS.
- J. Zhou, L. Guo, X. Guo, J. Mao and S. Zhang, Green Chem., 2010, 12, 1835–1843 RSC.
- G. D. Yadav, P. A. Chandan and D. P. Tekale, Ind. Eng. Chem. Res., 2011 DOI:10.1021/ie200446y.
- D. Y. Yoon, J. Park, H. Kang, P. S. Kim, I. Nam, G. K. Yeob, J. K. Kil and M. Cha, Appl. Catal., B, 2011, 101, 275–282 CrossRef CAS.
- A. Iglesias-Juez, A. B. Hungría, A. Martínez-Arias, A. Fuerte, M. Fernández-García, J. A. Anderson, J. C. Conesa and J. Soria, J. Catal., 2003, 217, 310–323 CAS.
- T. Nakatsuji, R. Yasukawa, K. Tabata, K. Ueda and M. Niwa, Appl. Catal., B, 1998, 17, 333–345 CrossRef CAS.
- X. She and M. Flytzani-Stephanopoulos, J. Catal., 2006, 237, 79–93 CrossRef CAS.
- M. F. L. Johnson and C. D. Keith,, J. Phys. Chem., 1963, 67, 200–201 CrossRef CAS.
- S. W. Weller and A. A. Montagna, J. Catal., 1971, 20, 394–407 CrossRef CAS.
- E. Ruckenstein and M. L. Malhotra, J. Catal., 1976, 41, 303–311 CrossRef CAS.
- E. Ruckenstein and Y. F. Chu, J. Catal., 1979, 59, 109–122 CrossRef CAS.
- E. Ruckenstein and S. H. Lee, J. Catal., 1984, 86, 457–463 CrossRef CAS.
- Z. Qu, W. Huang, M. Cheng and X. Bao, J. Phys. Chem. B, 2005, 109, 15842–15848 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |