Syntheses of cyclic carbonates with amidinium halide catalysts in reusable, reversible, room-temperature ionic liquids or acetonitrile†
Tao
Yu
and
Richard G.
Weiss
*
Department of Chemistry, Georgetown University Washington, D.C. 20057-1227, USA. E-mail: weissr@georgetown.edu
First published on 18th November 2011
Abstract
In situ-prepared, reversible, room-temperature ionic liquids (RTILs), amidinium carbamates, have been used as media for the syntheses of cyclic carbonates by cycloaddition of CO2 to epoxides. The amidinium carbamates were prepared by exposing equimolar mixtures of an easily synthesized amidine and a primary amine to CO2 gas. For comparison purposes, amidinium dithiocarbamates were also employed as the RTILs in some experiments. Reaction between CO2 and four epoxides in the RTILs, occurs in good yields (> 90% in many cases) at room temperature or 50 °C in the presence of an amidinium halide catalyst. Product and any unreacted epoxide were extracted easily upon addition of an immiscible, lower density liquid to an RTIL reaction mixture. This process was repeated three times with the same RTIL without any obvious decrease in catalytic activity; presumably, additional transformations could have been conducted. The influences of the type of catalyst, CO2 pressure, reaction time, and temperature on the reaction yields have been investigated. The relatively mild reaction conditions and ease of separation of products, as well as the ability of the amidinium carbamates to be reused in the presence or absence of water make this an attractive alternative to other procedures for the efficient syntheses of cyclic carbonates.
Introduction
Chemical fixation of CO2 and its use as an inexpensive, nontoxic, highly abundant, single carbon atom (C1 resource) building block are of great current interest due to the growing concern about the deleterious effects of greenhouse gases on our environment.1,2 The syntheses of five-membered ring carbonates from CO2 and epoxides (Scheme 1) is a promising methodology from the standpoint of resource utilization because this reaction has 100% atom economy and cyclic carbonate products are widely used for a variety of applications, such as polar aprotic solvents, precursors of polymeric materials, and intermediates in the syntheses of pharmaceuticals.3,4,5 Numerous catalysts, including zeolites, metal halides, metal complexes and Lewis acids or bases have been developed and investigated to facilitate the reaction between an epoxide and a molecule of CO2.6 However, problems associated with the separation of products from the solvents and catalysts after reaction and sensitivity of catalysts to water or air7 still need to be addressed. Also, the nature of the solvent can be a critical element in the ease of reaction.8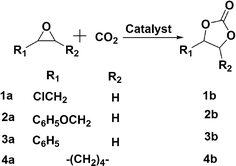 | ||
| Scheme 1 | ||
Although ionic liquids, by themselves, are not necessarily ‘greener’ than other solvents, they can offer intrinsic advantages in catalytic reactions over many other organic solvents in reactions involving CO2,9 especially in the steps leading to isolation of the products and to minimize byproducts such as polycarbonates10 or polyesters11 in reactions involving epoxides as co-reactants. In addition, high temperatures (>100 °C),12 high pressures of CO213 and co-catalyst systems14 are usually required to obtain good yields of cyclic carbonates from epoxides.
Previously, we developed a new class of thermally-reversible, room-temperature ionic liquids (RTILs).15,16,17 They are made from easily synthesized, readily available materials and can be transformed reversibly to their non-ionic liquid states. The non-ionic liquids consist of equimolar mixtures of an N′-alkyl-N,N-dimethylacetamidine (L) and an aliphatic amine (M). When exposed to 1 atm of CO2 gas, 1/1 (mole/mole) amidine/amine solutions become cationic–anionic pairs, amidinium carbamates (eqn (1)). Heating the ionic liquids in air to ca. 50 °C or bubbling N2 gas through them at ambient temperatures for protracted periods displaces the CO2 and re-establishes the non-ionic amidine/amine states. Utilizing similar chemistry, two classes of chiral RTILs16,17 were also prepared using a chiral amino acid ester or a chiral amino alcohol as the amine moiety. The reversibility of these achiral and chiral RTILs offers significant potential benefits as solvents in the syntheses and separations of products which are soluble in low-polarity media and whose mechanisms of formation depend on catalysts soluble only in high-polarity media. Unlike many other ILs, the ones employed here (as well as the catalysts derived from the amidines) can be exposed to water without deleterious consequences.
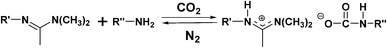 | (1) |
Here, a prototype reaction, the synthesis of cyclic carbonates by cycloaddition of CO2 to epoxides, utilizing the in situ-reversibility of the RTILs, is described (Scheme 1). The amidine (R′ = -(CH2)6H; C6) and amine components of the amidinium carbamate RTILs were prepared as described previously.15 For comparison purposes, amidinium dithiocarbamates, prepared by adding CS2 to 1/1 (mole/mole) amidine/amine solutions (eqn (2)), were employed as the RTILs18 in some experiments. Reaction between a number of epoxides and CO2 at 25–50 atm of pressure is found to occur in yields > 90% in the RTILs in many cases at ambient or slightly elevated temperatures in the presence of an amidinium halide or other Lewis acid/base catalyst; the yields are somewhat lower at both lower and higher CO2 pressures, but for very different reasons. The postulated mechanism for formation of the cyclic carbonates is shown in Scheme 2, The cyclic carbonate products and any unreacted epoxide could be extracted easily upon addition of an immiscible, lower density liquid (e.g., an alkane or ether) to an RTIL reaction mixture. Then, bubbling of CO2 through the amidinium carbamate (L-M-CO2) was sufficient to remove traces of the extracting liquid so that it and the catalyst could be reused. The influence of the type of catalyst, CO2 pressure, reaction time and temperature on the reaction yields are reported.
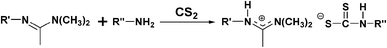
| (2) |
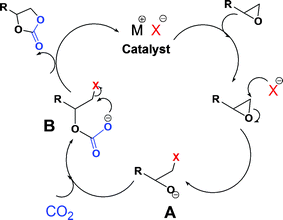 | ||
| Scheme 2 Postulated mechanism for catalyzed syntheses of cyclic carbonates from epoxides and CO2.19 | ||
The relatively mild reaction conditions, ability to use metal-free catalysts, ease of separation of products from the amidinium carbamate, and the ability to reuse the solvent and catalyst with little or no decrease of product yield make the procedures described here an attractive alternative to other methods for the syntheses of cyclic carbonates.
Experimental section
Materials
Epichlorohydrin (99%), 1,2-epoxy-3-phenoxypropane (99%), styrene oxide (97%), cyclohexene oxide (98%), tetraethylammonium bromide (98%) and lithium bromide (>99%) were purchased from Sigma–Aldrich and used as received. Carbon dioxide (CO2) was supplied by GT & S with a purity > 99.9%. Sodium bromide (99.9%), zinc chloride (99.2%)and zinc iodide (99.9%) were from Fisher. Zinc bromide (99.9%) was from Baker. Hydrochloric acid (HCl, 99%, 37 wt% in water), hydroiodic acid (HI, 99.95%, 57 wt% in water), and hydrobromic acid (HBr, 99.9%, 48 wt% in water) are from Baker. All organic solvents were HPLC grade (Fisher or Aldrich). Acetonitrile was dried over 4A molecular sieves before being used. Carbon disulfide (anhydrous, 99.9%), biphenyl (99.5%), n-hexylamine (99%) were purchased from Aldrich. Chloroform-D (99.8% D) was purchased from Cambridge Isotope Laboratory. Silica gel for column chromatography was purchased from YB in San Clemente, CA. The aliphatic amidines were available from previous studies.16Instrumentation
IR spectra were obtained on a Perkin-Elmer Spectrum One FTIR spectrometer interfaced to a PC, using an attenuated total reflection accessory or NaCl plates. NMR spectra were recorded on an Inova 400 MHz NMR spectrometer operating at 400 MHz (1H), or 100.5 MHz (13C), respectively. 1H and 13C NMR spectra were indirectly referenced to TMS using residual solvent signals as internal standards and CDCl3 as the solvent. GC-MS was performed on a Varian Saturn 2100 instrument in the electron ionization (EI) mode using a DB-5 (15 m × 0.25 mm) column.The high pressure stainless steel cell and the CO2 compressor apparatus are described in detail in Supporting Information (Fig. S1†). The temperature variation throughout the cell at a nominal temperature of 50 °C is estimated to be ca. 1.5 °C based upon measurements of the internal and external temperatures of the cell at various points.
General experimental procedure
Procedure for the syntheses of cyclic carbonates
A typical reaction procedure is described for the synthesis of 4-phenoxymethyl-1,3-dioxolan-2-one (2b) from CO2 and 1,2-epoxy-3-phenoxy propane (2a) (Scheme 1) using LiBr as a catalyst in the ionic liquid amidinium carbamate, C6-hexylamine-CO2. Organic salts, amidinium halides (Cn-HX), were used as catalysts as well. In all cases, 1H, 13C NMR and MS spectra of the products were consistent with those in the literature.20In an open glass vial with a 1 cm long Teflon stir bar, LiBr (1.2 mg, 1.4 × 10−2 mmol) was added to a solution of 1,2-epoxy-3-phenoxypropane (2a) (0.10 g, 0.70 mmol) in 0.50 mL of C6-hexylamine-CO2, prepared by bubbling CO2 through a C6/hexylamine mixture for 10 min. The glass vial was placed in the stainless-steel, high-pressure cell. Then, CO2 gas was introduced to a desired pressure and the cell temperature was raised. After stirring the reaction mixture for several hours, the pressure was brought slowly back to one atmosphere and diethyl ether (3.0 mL×3) was used to extract the products and any unreacted epoxide from the reaction mixture. NMR and GC-MS spectra of the product mixtures, after removing the diethyl ether, showed no discernible ionic liquid or its precursor amidine and amine components. In some cases, the products were isolated by combining the etherates and reducing them carefully to residue which was separated by column chromatography (silica, 50–75 μm) using 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 hexane:EtOAc as the eluent. Spectra of the isolated products are collected in Supporting Information.† Product yields were calculated from the weight of the residues (which were a combination of product and starting material only, whose proportion was calculated by integration of characteristic peaks in 1H NMR spectra, or (as indicated) by the weight of isolated product after column chromatography. Some reactions were repeated and the reproducibility of the yields was ± 3%. (GC response factor calculations are shown in Fig. S2 of Supporting Information.†)
:3 hexane:EtOAc as the eluent. Spectra of the isolated products are collected in Supporting Information.† Product yields were calculated from the weight of the residues (which were a combination of product and starting material only, whose proportion was calculated by integration of characteristic peaks in 1H NMR spectra, or (as indicated) by the weight of isolated product after column chromatography. Some reactions were repeated and the reproducibility of the yields was ± 3%. (GC response factor calculations are shown in Fig. S2 of Supporting Information.†)
Syntheses of amidinium halides
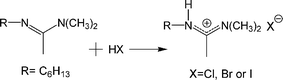
| (3) |
The C6-HX salts were prepared by the addition of an acid to C6 amidine (eqn (3)). A detailed example is presented for the synthesis of C6-HBr. C6-HCl and C6-HI were prepared in analogous fashions using 37% aqueous HCl and 57% aqueous HI, respectively. To a solution of C6 amidine (0.34 g, 2.0 mmol) in a stirred round bottom flask, 48% aqueous HBr (0.69 g, 4.0 mmol HBr) was added dropwise. After stirring the reaction mixture for 2 h at room temperature, most of the water was removed on a rotary evaporator. The residue was placed in a vacuum oven at 250 Torr and 50 °C for 48 h before characterization;21 see 1H NMR, 13C NMR, MS and IR spectra in Supporting Information†). Yield 0.79 g (95%) of a yellowish liquid; 1H NMR (CDCl3): 3.26 (t, 2H, -C![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 2–N
2–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) ); 3.07 (s, 6H, -N–(C3)2); 2.15 (s, 3H, -NC(C3)–N); 1.43 (m, 2H, -C2–CH2–N); 1.15–1.2 (m, 6H,CH3–(C2)3-); 0.72 (t, 3H, C
); 3.07 (s, 6H, -N–(C3)2); 2.15 (s, 3H, -NC(C3)–N); 1.43 (m, 2H, -C2–CH2–N); 1.15–1.2 (m, 6H,CH3–(C2)3-); 0.72 (t, 3H, C![[3 with combining low line]](https://www.rsc.org/images/entities/char_0033_0332.gif) ). 13C NMR (D2O): 164.56; 44.31; 40.57; 37.95; 30.57; 29.09; 25.26; 21.78; 13.92 ppm; MS (m/z 172 M−Cl+); IR: 1646 cm−1 (CN+).
). 13C NMR (D2O): 164.56; 44.31; 40.57; 37.95; 30.57; 29.09; 25.26; 21.78; 13.92 ppm; MS (m/z 172 M−Cl+); IR: 1646 cm−1 (CN+).
C6-HCl: Yield: 88% of a yellowish liquid; 1H NMR (CDCl3): 3.28 (t, 2H, -C2–N); 3.09 (s, 6H, -N–(C3)2); 2.16 (s, 3H, -NC(C3)–N); 1.45 (m, 2H, -C2–CH2–N); 1.15–1.23 (m, 6H, CH3–(C2)3-); 0.73 (t, 3H, C). 13C NMR (CDCl3): 164.73; 44.48; 40.67; 37.88; 30.68; 29.21; 25.32; 21.86; 13.99 ppm; MS (m/z 172 M−Br+); IR: 1646 cm−1 (CN+).
C6-HI: Yield: 85% of a dark orange color solid, mp 33.5–34.8 °C; 1H NMR (CDCl3): 3.29 (t, 2H, -C2–N); 3.10 (s, 6H, -N–(C3)2); 2.17 (s, 3H, -NC(C3)–N); 1.46 (m, 2H, -C2–CH2–N); 1.18–1.23 (m, 6H,CH3–(C2)3-); 0.74 (t, 3H, C). 13C NMR (CDCl3): 164.66; 44.49; 40.63; 37.82; 30.78; 29.28; 25.38; 21.88; 14.05 ppm; MS (m/z 172 M−I+); IR: 1646 cm−1 (CN+).
Solvent recycling test
The recycling procedure is illustrated in Scheme 3. The experimental steps in consecutive runs were the same as the initial run. Also, CO2 at 1 atm was bubbled into the RTIL solvent for 10 min after each run to ensure that it consisted of only amidinium carbamate and to remove any residual ether; according to the 1H NMR spectra, no ether remained after this procedure. Then, the same amount of 1,2-epoxy-3-phenoxypropane (2a) was added to the reaction mixture to start the next cycle.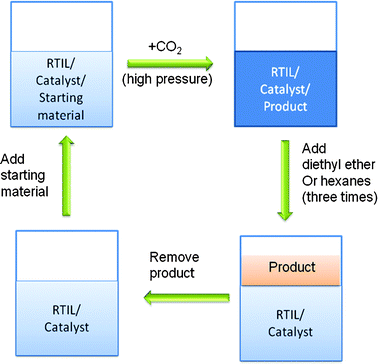 | ||
| Scheme 3 Diagram of the recycling procedure for reuse of RTIL/catalyst mixtures in the syntheses of cyclic carbonates from CO2 and an epoxide. | ||
Attempted polymerization of cyclohexene oxide
In CH 3 CN: In an open glass vial with a 1 cm Teflon stir bar, C6-HBr (10 mg, 4.0 × 10−2 mmol) was added to a solution of cyclohexene oxide (3a) (0.20 g, 20 mmol) in 0.25 mL of CH3CN. The glass vial was placed in the stainless steel high pressure cell under 25 atm CO2 and the temperature was increased to 50 °C. The work-up procedures are the same as described above.In solvent-free conditions: The procedure is the same as in CH3CN except that only cyclohexene oxide (3a) (0.80 g, 80 mmol) and C6-HBr (40 mg, 0.16 mmol) were employed.
Results and discussion
The postulated mechanism in Scheme 2 involves the ring opening of the epoxide by means of nucleophilic attack at the less hindered carbon atom by X− (as Br− in most of the reactions explored here) which leads to an oxy anion intermediate A. Then, CO2 reacts with A to form anion B which forms the carbonate products upon ring closure.22Three mono-substituted epoxides – epichlorohydrin (1a), 1,2-epoxy-3-phenoxypropane (2a), and styrene oxide (3a) – and one disubstituted epoxide, cyclohexene oxide (4a), have been tested as substrates. Both hexane and diethyl ether, solvents in which the epoxides and cyclic carbonates are very soluble, are almost completely immiscible with our RTILs.17 Therefore, they were selected to extract products from the RTILs in our experiments. The reactions were conducted between room temperature and 50 °C, a much lower temperature range than reported for the syntheses of cyclic carbonates from epoxides and CO2 in most procedures.6 However, the yields are lower than found in several other procedures to produce 2b.5 Also, Endo and coworkers reported recently that an amidine-mediated system, an N-methyltetrahydropyrimidine-CO2 adduct (MTHP-CO2), is capable of catalyzing cyclic carbonate syntheses at room temperature and 1 atm of CO2 pressure in the presence of a high concentration of LiBr catalyst (25 mol%).23,24
In an initial survey, the catalytic activities of various metal salt catalysts were evaluated in our RTILs (Table 1). When the anion is Br−, Li+ is the most efficient of the cations examined. Although Zn2+ is a stronger Lewis acid than Li+,25 its catalytic activity is lower here. The type of anion is important to the yield as well. Of the catalysts with Zn2+ as the cation, the one with Br− shows the highest catalytic activity. Polarity of the salts, their hard–soft/acidity–basicity,26 and their solubility in the RTILs may also affect the reaction yields. Overall, the trends in Table 1 are consistent with literature results—the lithium cation is expected to have the highest catalytic activity.27 Note that no reaction occurred in the absence of a catalyst; the RTIL does not serve as a catalyst. Also, no byproducts, such as polycarbonates or polyesters,28 were detected by GC-MS or by 1H NMR in the products isolated by column chromatography.
| Catalyst | Yield (%)a |
|---|---|
| a Yields by 1H NMR spectroscopy or isolated (in parentheses). | |
| LiBr | 70 (65) |
| Et4NBr | 62 |
| NaBr | 24 |
| ZnBr2 | 67 (64) |
| ZnCl2 | 45 |
| ZnI2 | 38 |
| (No catalyst) | 0 |
The yields of cycloaddition products using LiBr as catalyst and C6-hexylamine-CO2 as solvent (Table 2) are again, somewhat lower than those reported using other reaction conditions.29 At least in part, they can be attributed to the slower diffusion of bromide anion and other species in the RTILs (due to the relatively high local viscosity of RTILs, especially at room temperature), and the higher probability that intermediates A lose the X group than reacting with CO2 to form the intermediates B (Scheme 2). Tetra-alkylammonium bromide salts, including the tetramethyl, tetraethyl and tetrapropyl, have also been investigated as catalysts supported on silica (without solvent).30 All produced ≥ 96% yields of cyclic carbonates 1b, 2b and 3b.
| Epoxide | Product | Yield (%)a |
|---|---|---|
| a Yields were calculated by 1H NMR spectroscopy. Isolated yields are shown in parentheses. | ||
| 1a | 1b | 75 |
| 2a | 2b | 85 (84) |
| 3a | 3b | 82 (79) |
| 4a | 4b | 80 |
To extend further the versatility of this reaction, we explored the use of in situ prepared organic salts, amidinium halides (Cn-HX) to catalyze the reaction. This preparation is convenient for conducting the reactions and results in much higher yields (by ∼15%) of the products from 1a–4a than when metal halides are employed (Table 3). The enhancement of catalyst performance is attributed to the ability of the amidine itself to capture CO2 molecules23 and deliver them to the reactive site in a catalytic cycle (Scheme 2). Although the pKa's of the protonated amidines used here are not known, the pKa of protonated 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) is ca. 12.31 Assuming reasonably a similar pKa for our protonated amidines, they may form a hydrogen bond with the oxygen atom of an epoxide, thereby activating it for nucleophilic attack during the reaction. However, 1H NMR spectra show no chemical shift change of protons after mixing C6-HX with styrene oxide.32 Note also that commercially available amidines, such as DBU, can be used as amidinium catalysts. As shown in Table 4, C6-HBr and C6-HI are better catalysts than C6-HCl in this system, indicating the importance of nucleophilicity on the reaction rate.33 Given the estimated experimental error of the measurement of product yields (± 3%), it is difficult to differentiate the catalytic activities of C6-HBr and C6-HI.
In addition, the amidinium halide, prepared by addition of C6 to an equimolar amount of an HX (X = Cl, Br, and I) as a concentrated aqueous solution, was used as the catalyst for the conversion of 2a to 2b (Table 4). The yields of 2b with ‘wet’ C6-HX were ca. 8–11% lower than with dry C6-HX, but were still > 80%; amidinium carbamate remains an effective medium and the C6-HX are effective catalysts even in the presence of water. Within experimental error, ± 3%, water has about the same impact on the catalytic activities of each of the C6-HX. Also, the trend of the ‘wet’ Cn-HX catalytic activities are the same as those of the dry catalysts: C6-HI or C6-HBr is a better catalyst than C6-HCl.
Although the presence of 3 wt% H2O in a C6/leucine methyl ester mixture is known not to impede formation of the amidinium carbamate upon exposure to 1 atm of CO2,16 reaction between wet DBU and CO2 produces the amidinium bicarbonate.34 Analogous reactions, resulting in amidinium bicarbonate salts from a Cn amidine and water, must occur. Bicarbonate, rather than CO2, may be involved in the formation of the cyclic carbonates and it has been reported that 0.3 equivalents of water promoted the syntheses of cyclic carbonates from CO2 and epoxides.35 Also, a molecule of water and the anion of a Lewis base (PPh3BuI) play a synergistic role in opening an epoxy ring and in stabilizing the carbonate intermediates:35 at 125 °C under 20 atm of CO2 for 1 h, the yields of cyclic carbonate from 3a were 95% in the presence of water and only 25% in its absence.
The influence of CO2 pressure on reaction yields were compared using LiBr and C6-HBr as catalysts (Table 5). The yields of cyclic carbonate 2b increased with increasing pressure from 1 to 25 atm CO2. However, the yields decreased as pressure was increased further to 60 atm. Similar trends have been found for cyclic carbonate syntheses in other media and explained as follows:36 the initial increasing yields as pressure is changed from 1 to 25 atm is attributed to increasing CO2 concentrations within the RTIL solvent; the decreasing yields yet higher pressures are presumably a consequence of dilution of the substrate and catalyst as they are partitioned between the RTIL and a pre-supercritical phase of CO237 that moves some of the reactant into the total volume of the stainless steel cell (i.e., a gas-like phase).38,39 In so doing, the concentration of epoxide in the vicinity of the catalyst (which is retained within the original liquid volume) is lowered, resulting in lower product yields. In the experiments conducted here, a small amount of liquid was observed outside the glass vial after the reactions, especially in runs conducted at higher CO2 pressures.
As expected, longer reaction times and higher temperatures resulted in increased yields of cyclic carbonate. For example, the yield of cyclic carbonate 3b during the first 5 h increased rapidly and gradually reached a plateau value after 12 h (Fig. 1). The specific reasons for the higher yields with C6-HBr than with LiBr were not investigated here. They may involve the relative solubilities of the two catalysts and how they are able to interact with the key intermediates A and B in Scheme 2. Similarly, the increased yields of cyclic carbonate 3b at higher temperatures (Fig. 2) are attributed to the lowered viscosity of the RTIL as well as an intrinsic rise in the rates of all the steps leading to product. Lower viscosity causes higher rates of diffusion of cations and ions, allowing more efficient capture of CO2 by the critical intermediate A shown in Scheme 2 by making less probable the loss of the nucleophile (Xgroup) and reversion to epoxide.
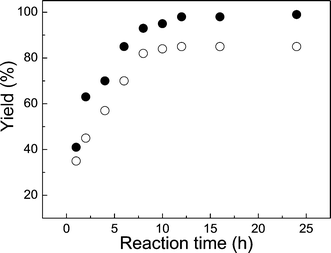 | ||
| Fig. 1 Influence of reaction time on the yield of cyclic carbonate 3b from 1.40 mol L−13a in C6-hexylamine-CO2 at 50 °C and under 25 atm of CO2 using 2 mol% of LiBr (○) or C6-HBr (●) as catalyst. Yields were calculated by 1H NMR spectroscopy. | ||
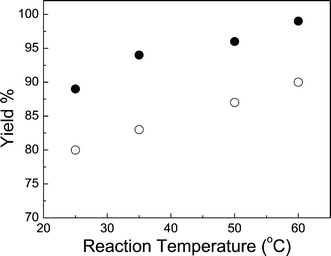 | ||
| Fig. 2 Influence of temperature on the yield of cyclic carbonate 3b from 1.40 mol L−13a after 8 h, 25 atm of CO2 in C6-hexylamine-CO2 using 2 mol% of LiBr (○) or C6-HBr (●) as catalyst. Yields were calculated by 1H NMR spectroscopy. | ||
Amidinium dithiocarbamate RTILs, prepared by adding CS2 to amidine/amine mixtures, usually have higher viscosities and better thermal stabilities than their CO2 analogues, amidinium carbamates.18 The differences between the polarities of amidinium carbamate and amidinium dithiocarbamates are very small,17,18 and apparently do not play an important role here. However, viscosity is important because lower viscosities increase the probability that intermediates A in Scheme 2 will encounter (and react with) a molecule of CO2 rather than losing the Xgroup and returning to the epoxide. Thus, the amidinium carbamate solvent give higher product yields (Table 6).
| Epoxide | Product | Yield (%) (A)a | Yield (%) (B)a |
|---|---|---|---|
| a Yields were calculated by 1H NMR spectroscopy. | |||
| 1a | 1b | 95 | 75 |
| 2a | 2b | 97 | 82 |
| 3a | 3b | 97 | 77 |
| 4a | 4b | 93 | 72 |
Furthermore, the recyclability of the RTIL/catalytic system under the optimum condition using the procedures outlined in Scheme 3 was explored. The results in Table 7 show that the efficiency of the LiBr catalyzed reactions decreased very slightly upon reuse, but still remained effective after 3 runs. Because lithium salts have a high affinity for carbonates40 and are sparingly soluble in ether, some LiBr catalyst may have been lost during product separation (i.e., repeated extraction with diethyl ether) and be responsible for the small decreases in yields in the second and third runs. Although there is a trend toward slightly lower yields with C6-HBr as catalyst, all of them are very high and within the limits of experimental error, estimated to be ±3%.
| Run | Yield (%) (A)a | Yield (%) (B)a |
|---|---|---|
| a Yields were calculated by 1H NMR spectroscopy. | ||
| 1 | 85 | 98 |
| 2 | 82 | 95 |
| 3 | 78 | 94 |
Jessop and coworkers41 reported a switchable ionic liquid prepared from a secondary amine and CO2, with Cr(salen)Cl as a catalyst that could be reused for the copolymerization of cyclohexene oxide (2a) and CO2, but that the yield of polycarbonate was reduced by ca. 20% during the second run and the polydispersity increased from 1.06 to 1.10. By contrast, reaction of 2a and 25 atm CO2 at 50 °C using C6-HBr as a catalyst in CH3CN or under solvent-free conditions for 24 h led only to 2b in 55 (CH3CN) or 42% yield (neat conditions); FT-IR and 1H NMR spectra showed no peaks attributable to a polycarbonate.10a In the latter case, the epoxide, as well as the resulting cyclic carbonate, act as the solvent.42 The lack of polymer as a competing product suggests that very high substrate concentrations may be possible without sacrificing the ‘cleanliness’ of reaction when amidinium bromides are used as the catalysts, and increasing the reaction time to 72 h and the temperature to 80 °C increased the reaction yield of 2b to ∼99%. Again, no evidence for polymer was found. We conjecture that the rate of ring closure to cyclic carbonate by intermediate B in Scheme 2 (when generated by amidinium bromide) must be much faster than the rate at which a second molecule of epoxide can intervene.
A zeolite-based, organic–inorganic hybrid catalyst has been reported to give 86–100% yields of cyclic carbonate 2b from neat 2a under 6.8 atm of CO2 and at 120 °C for 8 h.43 Also, bis(triphenylphosphine)immium (PPN) salts, [PPN]+Cl−, and PPN-manganese carbonylates, [PPN]+[Mn(CO)4L]− (L = CO, PPh3), have been found to be good catalysts for cyclic carbonate syntheses under solvent-free conditions. No polycarbonate was produced in these reactions when they were conducted under 5 atm of CO2 and at 100 °C.44 The relatively low solubility of CO2 in conventional solvents42 is a primary motivation to search for alternatives to the reaction between CO2 and epoxides.45 Supercritical CO2 has also been found to be an effective solvent for cyclic carbonate formation, although very high pressures are necessary.46 For example, in the presence of a Re(CO)5Br catalyst, the reaction of epoxides with supercritical CO2 without solvent at 110 °C afforded good yields.47 Thus, our catalyst appears to be competitive with all of the others reported to date for the cycloadditions, and it is superior to most in terms of cost and required temperature.
Conclusions
We have described the syntheses of five-membered cyclic carbonates by the addition of CO2 to epoxides using amidinium halides and other catalysts in a reversible room-temperature ionic liquid or acetonitrile. The conditions for performing the reactions are relatively mild when compared with most of the other methods in the literature, the yields are generally >90% at CO2 pressures of several atmospheres, the products are easily separated from the reaction mixtures, and the RTIL/catalyst systems can be reused with the same or different epoxide substrates. Only the desired cyclic carbonates were produced upon reaction of the four epoxide substrates examined; the transformations are very clean under the reaction conditions. Notably, no metal catalyst is required to effect the reaction in our RTIL systems. Because the RTIL and catalysts can be reused and the temperatures and required CO2 pressures are lower than in most other solvent/catalyst systems reported in the literature6—even 1 atm of CO2 and room temperature lead to some product—this approach is an attractive alternative for preparing five-membered heterocycles and CO2 fixation.Future work will be directed to additional exploitation of these and other RTILs16,17 developed by our group in the same and different reactions. For example, the RTILs may be able to effect syntheses of oxazolidinones from aziridines and CO2,48,49 and to prepare polycarbonates from epoxides and CO210 (under somewhat different experimental conditions than employed here). Also, we50 are developing polymer-supported6g,51 and optically-active amidinium halides as reusable catalysts to facilitate further the separation of products from the reaction media and to make chiral cyclic carbonates and oxazolidinones.52,53
Acknowledgements
We thank the U. S. National Science Foundation for its financial support of this research. We are grateful to Prof. Rodrigo Cristiano, Mr. William Craig and Mr. Leon Der for their help in assembling and designing parts of the high pressure apparatus. We are very grateful to Profs. Pedro F. Aramendía and Laura Japas of the University of Buenos Aires (Argentina) for designing and constructing the stainless steel reaction cell and for invaluable technical advice. Prof. Christian Wolf and Mr. Peng Zhang are thanked for the use of their chiral column and HPLC.Notes and References
- (a) D. M. D'Alessandro, B. Smit and J. R. Long, Angew. Chem., Int. Ed., 2010, 49, 6058–6082 CrossRef CAS; (b) C. A. Eckert, C. L. Liotta, D. Bush, J. S. Brown and J. P. Hallett, J. Phys. Chem. B, 2004, 108, 18108–18118 CrossRef CAS; (c) M. Wei, G. T. Musie, D. H. Busch and B. Subramaniam, J. Am. Chem. Soc., 2002, 124, 2513–2517 CrossRef CAS; (d) F. Jutz, J.-M. Andanson and A. Baiker, Chem. Rev., 2011, 111, 322–353 CrossRef CAS; (e) S. N. Riduan and Y. Zhang, Dalton Trans., 2010, 39, 3347–3357 RSC; (f) W. Leitner, Coord. Chem. Rev., 1996, 153, 257–284 CrossRef CAS; (g) J. Lu, M. J. Lazzaroni, J. P. Hallett, A. S. Bommarius, C. L. Liotta and C. A. Eckert, Ind. Eng. Chem. Res., 2004, 43, 1586–1590 CrossRef; (h) T. Yu, R. Cristiano and R. G. Weiss, Chem. Soc. Rev., 2010, 39, 1435–1447 RSC; (i) D. J. Darensbourg, Inorg. Chem., 2010, 49, 10765–10780 CrossRef CAS.
- M. M. Maroto-Valer, Ed. Carbon dioxide (CO2) capture transport and industrial applicationsWoodhead Pub., Ltd:Oxford, 2010 Search PubMed.
- (a) A. Baba, H. Kashiwagi and H. Matsuda, Organometallics, 1987, 6, 137–140 CrossRef CAS; (b) A. Baba, H. Kashiwagi and H. Matsuda, Tetrahedron Lett., 1985, 26, 1323–1324 CrossRef CAS.
- T. Sakakura, J-C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365–2387 CrossRef CAS.
- D. J. Darensbourg, S. J. Lewis, J. L. Rodgers and J. C. Yarbrough, Inorg. Chem., 2003, 42, 581–589 CrossRef CAS.
- (a) R. L. Paddock and S. T. Nguyen, J. Am. Chem. Soc., 2001, 123, 11498–11499 CrossRef CAS; (b) D. J. Darensbourg, C. C. Fang and J. L. Rodgers, Organometallics, 2004, 23, 924–927 CrossRef CAS; (c) Y. M. Shen, W. L. Duan and M. Shi, J. Org. Chem., 2003, 68, 1559–1562 CrossRef CAS; (d) H. W. Jing, S. K. Edulji, J. M. Gibbs, C. L. Stern, H. Y. Zhou and S. T. Nguyen, Inorg. Chem., 2004, 43, 4315–4325 CrossRef; (e) S. Liang, H. Liu, T. Jiang, J. Song, G. Yang and B. X. Han, Chem. Commun., 2011, 47, 2131–2133 RSC; (f) W. Clegg, R. W. Harrington, M. North and R. Pasquale, Chem.–Eur. J., 2010, 16, 6828–6843 CAS; (g) Y. Xie, Z. Zhang, T. Jiang, J. He, B. Han, T. Wu and K. Ding, Angew. Chem., Int. Ed., 2007, 46, 7255–7258 CrossRef CAS; (h) M. Tu and R. J. Davis, J. Catal., 2001, 199, 85–91 CrossRef CAS.
- (a) M. Kihara, N. Hara and T. Endo, J. Org. Chem., 1993, 58, 6198–6202 CrossRef CAS; (b) J. M. Sun, S. I. Fujita, F. Y. Zhao and M. Arai, Green Chem., 2004, 6, 613–615 RSC.
- (a) M. Aresta, A. Dibenedetto, L. Gianfrate and C. Pastore, J. Mol. Catal. A: Chem., 2003, 204, 245–252 CrossRef; (b) R. Srivastava, D. Srinivas and P. Ratnasamy, J. Catal., 2005, 233, 1–15 CrossRef CAS.
- (a) H. Kawanami, A. Sasaki, K. Matsui and Y. Ikushima, Chem. Commun., 2003, 896–897 RSC; (b) A. L. Zhu, T. Jiang, B. X. Han, J. C. Zhang, Y. Xie and X. M. Ma, Green Chem., 2007, 9, 169–172 RSC; (c) H. Zhou, W.-Z. Zhang, C.-H. Liu, J.-P. Qu and X.-B. Lu, J. Org. Chem., 2008, 73, 8039–8044 CrossRef CAS; (d) J.-L. Jiang, F. Gao, R. Hua and X. Qiu, J. Org. Chem., 2005, 40, 381–383 Search PubMed; (e) S.-F. Yin and S. Shimada, Chem. Commun., 2009, 1136–1138 RSC; (f) W. J. Kruper and D. V. Dellar, J. Org. Chem., 1995, 60, 725–727 CrossRef CAS; (g) Z. Yang, L.-N. He, C. Miao and S. Chanfreaua, Adv. Synth. Catal., 2010, 352, 2233–2240 CrossRef CAS.
- (a) D. J. Darensbourg, R. M. Mackiewicz, A. L. Phelps and D. R. Billodeaux, Acc. Chem. Res., 2004, 37, 836–844 CrossRef CAS; (b) D. J. Darensbourg, M. W. Holtcamp, G. E. Struck, M. S. Zimmer, S. A. Niezgoda, P. Rainey, J. B. Robertson, J. D. Draper and J. H. Reibenspies, J. Am. Chem. Soc., 1999, 121, 107–116 CrossRef CAS; (c) D. R. Moore, M. Cheng, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2003, 125, 11911–11924 CrossRef CAS; (d) X.-B. Lu and Y. Wang, Angew. Chem., Int. Ed., 2004, 43, 3574–3577 CrossRef CAS; (e) S. D. Thorat, P. J. Phillips, V. Semenov and A Gakh, J. Appl. Polym. Sci., 2003, 89, 1163–1176 CrossRef CAS; (f) D. J. Darensbourg, A. I. Moncada, W. Choi and J. H. Reibenspies, J. Am. Chem. Soc., 2008, 130, 6523–6533 CrossRef CAS.
- F. Jutz, J-M. Andanson and A. Baiker, Chem. Rev., 2011, 111, 322–353 CrossRef CAS.
- (a) K. Yamaguchi, K. Ebitani, T. Yoshida, H. Yoshida and K. Kaneda, J. Am. Chem. Soc., 1999, 121, 4526–4527 CrossRef CAS; (b) H. Kawanami, A. Sasaki, K. Matsui and Y. Ikushima, Chem. Commun., 2003, 896–897 RSC.
- (a) S. Zhang, Y. Chen, F. Li, X. Lu, W. Dai and R. Mori, Catal. Today, 2006, 115, 61–69 CrossRef CAS; (b) Y. X. Zhou, S. Q. Hu, X. M. Ma, S. G. Liang, T. Jiang and B. X. Han, J. Mol. Catal. A: Chem., 2008, 284, 52–57 CrossRef CAS.
- (a) F. Ono, K. Qiao, D. Tomida and C. Yokoyama, Appl. Catal., A, 2007, 333, 107–113 CrossRef CAS; (b) S.-S. Wu, X.-W. Zhang, W.-L. Dai, S.-F. Yin, W.-S. Li, Y.-Q. Ren and C.-T. Au, Appl. Catal., A, 2008, 341, 106–111 CrossRef CAS.
- T. Yamada, P. J. Lukac, M. George and R. G. Weiss, Chem. Mater., 2007, 19, 967–969 CrossRef CAS.
- T. Yamada, P. J. Lukac, T. Yu and R. G. Weiss, Chem. Mater., 2007, 19, 4761–4768 CrossRef CAS.
- T. Yu, T. Yamada, G. C. Gaviola and R. G. Weiss, Chem. Mater., 2008, 20, 5337–5344 CrossRef CAS.
- T. Yu, T. Yamada and R. G. Weiss, Chem. Mater., 2010, 22, 5492–5499 Search PubMed.
- (a) D. J. Darensbourg and M. W. Holtcamp, Coord. Chem. Rev., 1996, 153, 155–174 CrossRef CAS; (b) J. Sun, S. Zhang, W. Cheng and J. Ren, Tetrahedron Lett., 2008, 49, 3588–3591 CrossRef CAS.
- Y. Tsutsumi, K. Yamakawa, M. Yoshida, T. Ema and T. Sakai, Org. Lett., 2010, 12, 5728–5731 CrossRef CAS.
- In some reactions, the ‘wet’ (undried) catalysts were employed as well.
- V. Calo, A. Nacci, A. Monopoli and A. Fanizzi, Org. Lett., 2002, 4, 2561–2563 CrossRef CAS.
- B. Barkakaty, K. Morino, A. Sudo and T. Endo, Green Chem., 2010, 12, 42–44 RSC.
- Under the reaction conditions described in ref. 23 – 1 atm pressure of CO2 and room temperature for 24 h – but with C6 amidine as the solvent, a ca. 22% yield of cyclic carbonate was obtained from cyclohexene oxide. Also, under the same conditions but with 25 mol% C6-HBr as catalyst in C6 amidine or DMF as solvent, the yields of cyclic carbonate were 28% or 20%, respectively. Experimental details are reported in the Supporting Information†.
- J. March, Advanced Organic Chemistry, 4th ed., J. Wiley and Sons:New York, 1992 Search PubMed.
- (a) R. G. Pearson, J. Am. Chem. Soc., 1963, 85, 3533–3543 CrossRef CAS; (b) P. K. Chattaraj, H. Lee and R. G. Parr, J. Am. Chem. Soc., 1991, 113, 1855–1856 CrossRef CAS.
- S-I. Fujita, M. Nishiura and M. Arai, Catal. Lett., 2010, 135, 263–268 Search PubMed.
- X-B. Lu, Y-J. Zhang, B. Liang, X. Li and H. Wang, J. Mol. Catal. A: Chem., 2004, 210, 31–34 CrossRef CAS.
- (a) M. Yoshida, M. Fujita, T. Ishii and M. Ihara, J. Am. Chem. Soc., 2003, 125, 4874–4881 CrossRef CAS; (b) M. Yoshida and M. Ihara, Chem.–Eur. J., 2004, 10, 2886–2893 CrossRef CAS; (c) S. Fukuoka, M. Kawamura, K. Komiya, M. Tojo, H. Hachiya, K. Hasegawa, M. Aminaka, H. Okamoto, I. Fukawa and S. Konno, Green Chem., 2003, 5, 497–507 RSC.
- J.-Q. Wang, D.-L. Kong, J.-Y. Chen, F. Cai and L.-N. He, J. Mol. Catal. A: Chem., 2006, 249, 143–148 CrossRef CAS.
- D. A. Evans, pKa's of inorganic and oxoacids. http://evans.harvard.edu/pdf/evans_pKa_table.pdf.
- Three 1H NMR spectra were recorded: (1) 5.0 ×10−3 mol L−1C6-HI in CDCl3; (2) 0.5 × 10−3 M styrene oxide in CDCl3; (3) 5.0 × 10−3 M C6-HI and 0.5 × 10−3 M styrene oxide in CDCl3. The chemical shifts of the epoxide ring protons were carefully compared and no changes in the shifts were observed.
- (a) V. Calo, A. Nacci, L. Lopez and A. Napola, Tetrahedron Lett., 2001, 42, 4701–4703 CrossRef CAS; (b) V. Calo, A. Nacci, A. Monopoli, L. Lopez and A. Di Cosmo, Tetrahedron, 2001, 57, 6071–6077 CrossRef CAS.
- D. J. Heldebrant, P. G. Jessop, C. A. Thomas, C. A. Eckert and C. L. Liotta, J. Org. Chem., 2005, 70, 5335–5338 CrossRef CAS.
- J. Sun, J. Ren, S. Zhang and W. Cheng, Tetrahedron Lett., 2009, 50, 423–426 CrossRef CAS.
- (a) M. North, R. Pasquale and C. Young, Green Chem., 2010, 12, 1514–1539 RSC; (b) J. Song, Z. Zhang, S. Hu, T. Wu, T. Jiang and B. Han, Green Chem., 2009, 11, 1031–1036 RSC.
- E. J. Beckman, J. Supercrit. Fluids, 2004, 28, 121–191 CrossRef CAS.
- J. Sun, S-I. Fujita, F. Zhao and M. Arai, Green Chem., 2004, 6, 613–616 RSC.
- H. F. Zhang, B. X. Han, Z. S. Hou and Z. M Liu, Fluid Phase Equilib., 2001, 179, 131–138 CrossRef CAS.
- A. M. Belostotskii, E. Markevich and D. Aurbach, J. Coord. Chem., 2004, 57, 1047–1056 CrossRef CAS.
- L. Phan, J. R. Andreatta, L. K. Horvey, C. F. Edie, A.-L. Luco, A. Mirchandani, D. J. Darensbourg and P. G. Jessop, J. Org. Chem., 2008, 73, 127–132 CrossRef CAS.
- T. Sakakura, J.-C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365–2387 CrossRef CAS.
- R. Srivastava, D. Srinivas and P. Ratnasamy, Appl. Catal., A, 2005, 289, 128–134 CrossRef CAS.
- W. Sit, S. Ng, K. Kwong and C. Lau, J. Org. Chem., 2005, 70, 8583–8586 CrossRef CAS.
- T. Sakai, Y. Tsutsumi and T. Ema, Green Chem., 2008, 10, 337–341 RSC.
- G. W. Coates and D. R. Moore, Angew. Chem., Int. Ed., 2004, 43, 6618–6639 CrossRef CAS.
- J.-Li. Jiang, F. Gao, R. Hua and X. Qiu, J. Org. Chem., 2005, 70, 381–383 CrossRef CAS.
- (a) A. W. Miller and S. T. Nguyen, Org. Lett., 2004, 6, 2301–2304 CrossRef CAS; (b) Y.-M. Shen, W.-L. Duan and M. Shi, Eur. J. Org. Chem., 2004, 14, 3080–3089 CrossRef; (c) O. Ihata, Y. Kayaki and T. Ikariya, Angew. Chem., Int. Ed., 2004, 43, 717–719 CrossRef CAS; (d) Y. Du, Y. Wu, A.-H. Liu and L.-N. He, J. Org. Chem., 2008, 73, 4709–4712 CrossRef CAS.
- (a) Z.-Z. Yang, L.-N. He, S.-Y. Peng and A.-H. Liu, Green Chem., 2010, 12, 1850–1855 RSC; (b) W.-Z. Li, and R. G. Weiss, unpublished results.
- T. W. Farnsworth, T. Yu, and R. G. Weiss, unpublished results.
- C. Qi, J. Ye, W. Zeng and H. Jiang, Adv. Synth. Catal., 2010, 352, 1925–1933 Search PubMed.
- (a) J. T. Groves and R. S. Myers, J. Am. Chem. Soc., 1983, 107, 5791–5796 Search PubMed; (b) E. N. Jacobsen, Acc. Chem. Res., 2000, 33, 421–431 CrossRef CAS; (c) L. E. Martinez, J. L. Leighton, D. H. Carsten and E. N. Jacobsen, J. Am. Chem. Soc., 1995, 117, 5897–5898 CrossRef CAS; (d) E. N. Jacobsen, F. Kakiuchi, R. G. Konsler, J. F. Larrow and M. Tokunaga, Tetrahedron Lett., 1997, 38, 773–776 CrossRef CAS.
- Preliminary attempts to synthesize optically active cyclic carbonates using a chiral amidinium bromide catalyst, Nor-HBr, in CH3CN have not been successful, thus far. The epoxides, 3a and 4a, produced high chemical yields of the corresponding cyclic carbonates, 3b and 4b, but neither with a discernible enantiomeric excess.
.
Footnote |
| † Electronic supplementary information (ESI) available: Photographs of the high pressure CO2 generator, reaction cell and temperature control setup, diagram of the layout of the high pressure instrument components, GC response factor calculations, 1H NMR, 13C NMR, FT-IR, and MS spectra of cyclic carbonates and amidinium halide catalysts. See DOI: 10.1039/c1gc16027c |
| This journal is © The Royal Society of Chemistry 2012 |