A simple direct phosgeneless route to N-heteroaryl unsymmetrical ureas
Marianna
Carafa
a,
Valentina
Mele
a and
Eugenio
Quaranta
*abc
aDipartimento di Chimica, Università degli Studi di Bari “Aldo Moro”, Campus Universitario, Via E. Orabona, 4, 70126, Bari, Italy. E-mail: quaranta@chimica.uniba.it; Fax: (+39)-080-544-2129
bICCOM-CNR, Dipartimento di Chimica, Campus Universitario, Via E. Orabona, 4, 70126, Bari, Italy
cUdRBA1, Consorzio “INCA”, Via Delle Industrie, 21/8, 30175, Marghera (VE), Italy
First published on 23rd November 2011
Abstract
A new simple approach to the synthesis of unsymmetrical ureas HetNC(O)NRR′ (HetNH = pyrrole, indole, carbazole; R, R′ = H, alkyl, aryl) has been explored, which involves the direct reaction of the N-phenoxycarbonyl derivatives of pyrrole, indole and carbazole, HetNCO2Ph, with amines. The aminolysis reaction can be catalyzed by the amidine base DBU (1,8-diazabicyclo[5.4.0]undec-7-ene) under usually very mild conditions and provides a straightforward convenient entry into the target products through a route which avoids the traditional protocols based on multistep procedures and toxic phosgene or phosgene-derivatives.
Introduction
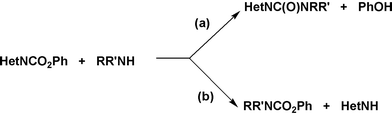
The search for new direct eco-friendly synthetic routes is a major challenge for modern chemistry.1 Despite the efforts currently devoted to redesign the synthesis of many classes of products according to strategies more sustainable from the environmental point of view, HetNC(O)NRR′ carboxamides of N-heteroarenes as pyrrole, indole and carbazole provide an example of chemicals which are still manufactured through traditional multistep methods based on risky and/or harmful starting materials. These particular unsymmetrical ureas,2,3 wherein one of the ureidic N atoms is also part of a heteroaromatic ring,4 are widely used as synthetic intermediates,5,6 find practical application in several fields,7 and usually exhibit biological activity or interesting pharmacological properties.8,9The classic methods of synthesis of these compounds start from derivatives of phosgene4,10 or directly from COCl2,5,8 a toxic and harmful species, the utilization of which in chemical synthesis finds, nowadays, larger and larger constraints due to governmental policies for environmental protection.11 The preliminary formation of a N-heteroaryl metal salt, HetNM, is often required. Usually, HetNC(O)NHR ureas are prepared by reaction of HetNM (M = Li, K, MgX) salts with isocyanates (Scheme 1(a)).10a,b,dN,N-Disubstituted ureas HetNC(O)NRR′4,5,8 have been often obtained by reaction of HetNM salts with N,N-disubstituted carbamoyl chlorides (Scheme 1(b)).4 A major additional problem of these approaches (Scheme 1) is also the regioselectivity of the electrophilic attack to the heterocyclic salt HetNM, which may depend on the nature of cation M and, therefore, necessitate the empirical characterization of the experimental conditions privileging N- over C-functionalization.12
 | ||
| Scheme 1 Classic phosgene-based synthetic routes to unsymmetrical ureas HetNC(O)NRR′. | ||
A relatively more recent method (Scheme 2) used CO2 as the source of carbonyl group and was based on the activation of carboxylic acid HetNCO2H (HetNH = pyrrole, indole) to anhydride [HetNC(O)]2O.13 However, the activation step requires the use of a stoichiometric amount of a coupling reagent as EDCI·HCl (1-[3-(dimethylamino)propyl]-3-ethylcarbodiimide hydrochloride), usually manufactured from phosgene.14 Moreover, the synthetic route is atomically uneconomical, as it implies a multistep procedure and needs 2 mol of HetNH (for preforming the anhydride) per mol of product.
 | ||
| Scheme 2 Synthesis of unsymmetrical ureas HetNC(O)NRR′via HetNCO2H activation. | ||
In the last few years many efforts have been addressed to develop a fully phosgene-free chemistry and the study of safe non-toxic active carbonyl species which can serve as substitutes for phosgene or phosgene-derivatives has been drawing growing attention.15–18 In this regard, carbonic acid diesters have been shown to play an important role15a,16–18 as these compounds are currently manufactured, even on the industrial scale, through phosgene-free routes.19,20 As a part of our studies in this field,16,17 we have recently reported on the direct carbonylation of N-heteroaromatics HetNH, such as pyrrole, indole and carbazole, with organic carbonates for the phosgeneless synthesis of carbonyl derivatives HetNCO2R,17b,d usually obtained through phosgenation methods.21 The carbonylation reaction can be catalyzed by superbases as DBU (1,8-diazabicyclo[5.4.0]undec-7-ene) or phosphazenes. Notably, the DBU-promoted reaction of HetNH (pyrrole, indole, carbazole) with diphenyl carbonate was shown to be an excellent new method for the selective high-yield synthesis of the N-phenoxycarbonyl derivatives HetNCO2Ph 1–3 [eqn (1)].17d Herein, with the intent of designing a fully phosgene-free green approach to the synthesis of unsymmetrical ureas HetNC(O)NRR′ (R, R′ = H, alkyl, aryl), we have explored the potential of compounds 1–3 as carbonylating agents of amines [eqn (2)]. Under usually mild conditions, the direct reaction of 1–3 with amines provided a straightforward phosgeneless access into N-heteroaryl unsymmetrical ureas HetNC(O)NRR′ (Fig. 1) through a convenient route which is an eco-friendly alternative to the classic methods described above.
 | (1) |
 | (2) |
 | ||
| Fig. 1 Synthesized unsymmetrical ureas. | ||
Results and discussion
A preliminary study concerned the reaction of a primary aliphatic amine such as benzylamine with 1-phenoxycarbonyl pyrrole (1), which was selected as the reference substrate (Table 1). At 293 K, in the absence of any catalyst, 1 reacted promptly with an excess of benzylamine, used both as reactant and solvent, to afford 1-benzylaminocarbonyl pyrrole (4) and phenol: under the used solventless conditions (entry 1, Table 1), 1 was quantitatively converted into 4 within a short time (1 h). The formation of 4 was very selective (>99%). The attack of the amine to the substrate caused the selective expulsion of phenoxy group with formation of 4, which, under the working conditions, did not react further with the amine to generate the symmetrical urea (RNH)2CO (R = benzyl). Accordingly, the IR spectrum of the reaction mixture in the range 1800–1600 cm−1 showed one only absorption at 1701 cm−1 due to 4.| Entry | PhCH2NH2 (mmol) | 1 (mmol) | DBU (mol%)a | Solvent | T/K | t/h | Conversionb [%] | 4 [%]c |
|---|---|---|---|---|---|---|---|---|
| a Relative to 1. b Of 1 (by GC). c Isolated yield (based on 1), unless otherwise specified. d 1 mL. e See also main text. f Selectivity to 4. g 2 mL. | ||||||||
| 1 | 5.95 | 1.07 | — | — | 293 | 1 | 100 | 86 |
| 2 | 0.66 | 0.59 | — | Diethyl ether d | 293 | 36e | ≈100 | >99f |
| 3 | 1.24 | 1.10 | 100 | Diethyl ether g | 293 | 2 | ≈100 | 87 |
| 4 | 1.11 | 1.08 | 9.9 | THF d | 338 | 2 | ≈100 | 90 |
Depending on the solvent properties and/or expensiveness of amine reactant, the use of solventless conditions, or a large excess of amine, may be poorly attractive from the synthetic point of view or not practicable at all. Therefore, the aminolysis reaction was studied also in ethereal solvents (diethyl ether or THF),22 using a modest excess of amine. In diethyl ether, at ambient temperature, the reaction of 1 with a slight excess (≈10 mol%) of PhCH2NH2 afforded 4 in practically quantitative yield (Entry 2, Table 1). However, under the used conditions, the conversion rate, while being satisfactory soon after mixing the reactants, became excessively slow in the long run (entry 2, Table 1) and the full conversion of the substrate required a markedly longer time.
The above behavior may reflect the fact that aminolysis of carbamates is usually catalyzed by amine itself, since a second molecule of amine can act as a catalyst of the process.23 We have, therefore, investigated the effect of a base organo-catalyst and focused the attention on the strong amidine superbase DBU. Under experimental conditions otherwise analogous to those used in entry 2 (Table 1), the presence of 1 equivalent of the amidine superbase enhanced markedly the conversion rate (entry 3, Table 1). The aminolysis reaction was promoted also by markedly lower loadings of DBU, but less effectively, unless the working temperature was increased. Accordingly, at 338 K, in the presence of 10 mol% of DBU, 1 converted quantitatively to 4 within only 2 h (entry 4, Table 1).
Table 2 summarizes the results obtained with a few other amines. Like benzylamine, at ambient temperature also allylamine reacted easily with 1 (RNH2/1 ≈ 1.14 mol/mol), in THF and in the presence of 1 equivalent of DBU, to afford 1-allylaminocarbonyl pyrrole (5) (entry 1, Table 2).
| Entry | amine (mmol) | 1 (mmol) | DBU (mol%)a | Solvent b | T/K | t/h | Conversionc [%] | Urea: [%]d |
|---|---|---|---|---|---|---|---|---|
| a Relative to 1. b 1 mL, when solvent (diethyl ether, THF) was used. c Of 1 (by GC). d Isolated yield (based on 1) of C4H4NC(O)NRR′, unless otherwise specified. e Selectivity. f The product formed in trace amounts. g After 8 days the conversion of 1 was close to 80%. h Not isolated. i After 4 days the conversion of 1 was close to 75%. j After 1 day the conversion of 1 was close to 70%. | ||||||||
| 1 | CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCH2NH2 (0.94) CHCH2NH2 (0.94) |
0.82 | 101 | THF | 293 | 2 | 100 | 5: 87 |
| 2 | O(CH2CH2)2NH (0.63) | 0.55 | 103 | diethyl ether | 293 | 2 | 100 | 6: >99e |
| 3 | O(CH2CH2)2NH (1.20) | 1.08 | 9.9 | THF | 338 | 2 | 100 | 6: 93 |
| 4 | (PhCH2)2NH (1.20) | 1.09 | 9.8 | THF | 338 | 6 | ≈ 0 | 7: f |
| 5 | (PhCH2)2NH (1.20) | 1.09 | 9.8 | THF | 393 | 285 | 100g | 7: h |
| 6 | (PhCH2)2NH (0.62) | 0.57 | 99.4 | THF | 393 | 166 | 99i | 7: h |
| 7 | (PhCH2)2NH (2.47) | 0.82 | 10.0 | — | 423 | 18 | 99 | 7: h |
| 8 | (PhCH2)2NH (2.47) | 0.82 | 102 | — | 423 | 5 | 100 | 7: h |
| 9 | (PhCH2)2NH (2.34) | 0.74 | 202 | — | 293 | 60 | 100j | 7: 81 |
| 10 | PhNH2 (5.49) | 0.55 | — | — | 293 | 18 | ≈0 | 8: f |
| 11 | PhNH2 (5.49) | 0.55 | 104 | diethyl ether | 293 | 48 | 99 | 8: 81 |
| 12 | PhNH2 (2.47) | 0.82 | 103 | — | 293 | 12 | ≈100 | 8: 83 |
| 13 | PhNMeH (2.40) | 0.81 | 108 | — | 293 | 38 | ≈100 | 9: 89 |
| 14 | PhNMeH (2.49) | 0.83 | 102 | — | 338 | 8 | 100 | 9: 88 |
| 15 | PhNMeH (2.77) | 0.85 | 10.2 | — | 373 | 48 | 99 | 9: 97e |
The behavior of a few representative secondary aliphatic amines was also studied. In ethereal solvents (diethyl ether, THF) the DBU-promoted aminolysis of 1 with a cyclic secondary amine such as morpholine proceeded smoothly under very mild conditions and produced the relevant unsymmetrical urea 6 selectively (≥99%) with quantitative yield (entries 2 and 3, Table 2). In comparison, a sterically encumbered secondary acyclic amine, such as dibenzylamine, was, by far, much less reactive. For instance, under conditions similar to those used for morpholine in entry 3 (Table 2), dibenzylamine exhibited very poor reactivity towards 1 (entry 4, Table 1). Even at 393 K, the DBU-assisted aminolysis of 1 with a very modest excess of (PhCH2)2NH (≈10 mol% vs.1), in THF, required prohibitively long reaction times (entries 5 and 6, Table 2). Much higher conversion rates, which depended on the used catalyst load (10–100 mol%), were observed at higher temperature (423 K) under solventless conditions, using the amine in larger excess with respect to the substrate (entry 7 and entry 8, Table 2). In the range 393–423 K (entries 5–8, Table 2) the selectivity towards 1-dibenzylaminocarbonyl pyrrole (7) was moderate (≈75–85%) because of side-formation of other species, such as, for instance, phenyl N,N-dibenzylcarbamate (the major by-product)24 and very minor amounts of 1,1′-carbonyldipyrrole.17d The formation of significant amounts of (PhCH2)2NCO2Ph demonstrates that, at these temperatures, the substitution of pyrryl moiety can seriously compete with that of phenoxy group. Higher selectivity to the target product 7 may be favored by markedly lower reaction temperatures. To overcome the consequent drawback due to the lower conversion rate a higher catalyst load was used. Accordingly, at 293 K, under solventless conditions, urea 7 was obtained selectively (≈99%) and quantitatively within a still reasonable reaction time (60 h) by reacting 1 with (PhCH2)2NH (amine/1 ≈ 3 mol/mol) in the presence 2 equivalent of DBU (entry 9, Table 2).
Under those conditions wherein benzylamine reacted readily with 1 in the absence of DBU (entry 1, Table 1), a primary aromatic amine such as aniline was practically inert towards 1 (entry 10, Table 2). The presence of DBU (1 equivalent vs.1) in the reaction mixture promoted the aminolysis of 1 with formation of 1-phenylaminocarbonyl pyrrole (8) even at ambient temperature (entries 11 and 12, Table 2). Under solventless conditions (entry 12, Table 2) the conversion rate was more satisfactory than in diethyl ether (entry 11, Table 2). The aminolysis reaction was promoted also by catalytic amounts of DBU, but, in this case, the quantitative conversion of 1 required a longer reaction time at ambient temperature. At 393 K (DBU = 10 mol% vs.1; PhNH2/1 = 3 mol/mol), the conversion was faster, but less selective because of major formation of N,N′-diphenylurea, which, under the working conditions, formed by further reaction of 8 with aniline.10a The analysis of the reaction mixture showed also the side-formation of trace amounts of 1,1′-carbonyldipyrrole.17d
The synthetic protocol followed for aniline was extended also to N-methyl aniline (entries 13–15, Table 2) for the synthesis of 1-(N-methyl-N-phenylaminocarbonyl) pyrrole (9). The aminolysis reaction was selective (>96%) and, for instance, even at the highest temperature investigated (373 K; entry 15, Table 2), the side-formation of MePhNCO2Ph was modest.
We have also explored the compatibility of the synthetic approach with the presence of a few functional groups in the molecule of amine and studied the ureidization of functionalized amines, such as the methyl ester of L-leucine, which we have used in the form of chlorohydrate salt, and a few amino alcohols (Table 3).
| Entry | amine reactanta (mmol) | 1 (mmol) | DBU (mol%)b | THF (mL) | T/K | t/h | Conversionc[%] | Urea: [%]d |
|---|---|---|---|---|---|---|---|---|
| a R is Me2CHCH2CH(CO2Me). b Relative to 1. c Of 1 (by GC). d Isolated yield (based on 1), unless otherwise specified. e No reaction was observed. f Corresponding to 1 equivalent of DBU with respect to the chlorohydrate salt. g Not isolated. h Corresponding to 1.97 equivalents of DBU with respect to the chlorohydrate salt. i Corresponding to 1.81 equivalents of DBU with respect to the chlorohydrate salt. j Selectivity to 11. k After 36 h the conversion was nearly quantitative and addition of more AE (0.011 mL, 0.20 mmol) caused the fast conversion of the residual amounts of 1. | ||||||||
| 1 | L-RNH3Cl (0.65) | 0.49 | — | 1 | 293 | 46 | e | 10: e |
| 2 | L-RNH3Cl (0.65) | 0.49 | 131f | 3 | 293 | 24 | <10 | 10: g |
| 3 | L-RNH3Cl (0.68) | 0.55 | 246h | 3 | 293 | 44 | ≈100 | 10: 84 |
| 4 | L-RNH3Cl (0.76) | 0.60 | 229i | 3 | 333 | 16 | ≈100 | 10: 87 |
| 5 | H2NCH2CH2OH (16.6) | 0.75 | — | — | 293 | 1 | 100 | 11: ≈100j |
| 6 | H2NCH2CH2OH (0.89) | 0.56 | — | 1 | 293 | 2 | ≈100 | 11: ≈100j |
| 7 | H2NCH2CH2OH (0.63) | 0.58 | — | 1 | 293 | 36 | k | 11: 85 |
| 8 | H2NCH2CH2OH (0.64) | 0.56 | 11.8 | 1 | 293 | 1 | 100 | 11: 81 |
| 9 | H2NCH2CH(Ph)OH (0.99) | 0.60 | — | 1 | 293 | 2 | 100 | 12: 91 |
| 10 | H2NCH2CH(Ph)OH (0.79) | 0.64 | 10.4 | 1 | 293 | 2 | ≈100 | 12: 80 |
| 11 | H2NCH2CH(Ph)OH (0.67) | 0.58 | 100.5 | 1 | 293 | 0.25 | 100 | 12: 78 |
At 293 K, the salt L-Me2CHCH2CH(CO2Me)NH3Cl (L-RNH3Cl) was poorly reactive towards 1 (entry 1, Table 3) because of poor nucleophilicity of quaternary N atom. A negligible conversion to urea 10 was observed also when the substrate and the salt were reacted at ambient temperature in the presence of 1 equivalent (vs the salt) of the amidine base (entry 2, Table 3), which, under the working conditions, acted mainly as a proton scavenger [eqn (3)]25 rather than as the catalyst. The aminolysis of 1 by the chlorohydrate salt required a larger amount of the amidine base and, under very mild temperature conditions (293–333 K), was achieved more effectively by working in the presence of ≈2 equivalents of DBU (vs the salt; entry 3 and entry 4, Table 3).28
| Me2CHCH2CH(CO2Me)NH3Cl + DBU ⇄ DBU·HCl + Me2CHCH2CH(CO2Me)NH2 | (3) |
In principle, the reaction of 1 with aminoalcohols H2N-R-OH may effect not only the functionalization of H2N-moiety but also that of OH group with formation of carbamate C4H4NC(O)O-R-NH2. Elsewhere, in fact, we have reported that 1 can react with alcohols through a transesterification reaction.17dMethanol reacted easily with 1, even at ambient temperature, to give 1-methoxycarbonyl pyrrole, C4H4NCO2Me, but more sterically crowded alcohols, as PhCH2OH or t-BuOH, required the use of higher temperatures and/or a catalyst as DBU. In the presence of DBU the transesterification process was less selective, as the amidine base can also cause the defunctionalization of the heteroaromatic ring with formation of pyrrole and carbonic acid diesters (ROC(O)OPh, (RO)2CO).17b,d
We have found that, in THF, in the presence of DBU (10-100 mol% vs.1), 1 reacted smoothly with aminoalcohols H2N-R-OH (H2N-R-OH/1 = 1.14–1.20 mol/mol) such as 2-aminoethanol (AE) or (±)-2-amino-1-phenylethanol (Ph-AE) to give, under very mild conditions (293 K), the corresponding ureas 11 and 12 with high yield (entries 8, 10 and 11, Table 3). However, variable amounts of C4H4NC(O)NHCH2CHRO(O)CNC4H4 (17: R = H; 18: R = Ph) also formed under the working conditions (see also Experimental).
Scheme 3 shows two possible routes for the formation of C4H4NC(O)NHCH2CHRO(O)CNC4H4, both of which, in principle, may be operative. However, we note that in no case we detected (by GC, GC-MS) the presence of C4H4NCO2CHRCH2NH2 (R = H, Ph) in the reaction mixture. This fact suggests that C4H4NCO2CHRCH2NH2, if it really formed, did not accumulate in the reaction mixture, but reacted fast to give C4H4NC(O)NHCH2CHRO(O)CNC4H4.
 | ||
| Scheme 3 Possible routes to C4H4NC(O)NHCH2CHRO(O)CNC4H4. | ||
The selectivity towards the target products can be controlled more easily by working in the absence of the amidine base and suitably varying the H2N-R-OH/1 molar ratio. For instance, only minor amounts of C4H4NC(O)NHCH2CH2O(O)CNC4H4 were detected in the reaction mixture when 1 was reacted with a slight excess of AE (AE/1 = 1.09 mol/mol; entry 7, Table 3), at 293 K, in THF. Under the above conditions most of substrate (≈90%) reacted within the first 4–5 h, but the conversion of the residual amounts of 1 required a longer time. The use of a higher H2N-R-OH/1 molar ratio (60–65 mol%; entry 6 and 9 in Table 3) or of solventless conditions (entry 5, Table 3) allowed to overcome the above drawback and to conjugate high selectivity (≈100%) towards 11 or 12 with more satisfactory conversion rate.
Table 4 shows a few examples of aminolysis of 1-phenoxycarbonyl indole (2) and 9-phenoxycarbonyl carbazole (3). Under the working conditions (Table 4), the aminolysis of 3 with morpholine proceeded more sluggishly. Moreover, substrate 3 reacted with the investigated amines (allylamine, morpholine) with moderate selectivity (≈70%) mainly because of competitive defunctionalization of 3 and formation of carbazole (Scheme 4, route (b)). In comparison, the analogous reactions of 1 (see also Table 2) and 2 with the same amines were more selective and only in the aminolysis of 2 with morpholine (entry 3, Table 4) minor amounts of HetNH (indole) and O(CH2CH2)2NCO2Ph were found in the reaction mixture. The above features may reflect the importance of steric factors (bulkiness of both the attacking amine and HetN group in the substrate) in controlling not only the rate of the aminolysis process, but also the direction of the substitution reaction.
| Entry | Amine (mmol) | HetNCO2Ph (mmol) | Urea: [%]b |
|---|---|---|---|
| a Solvent: THF (1 mL); DBU: ≈100 mol% vs.2 or 3; temperature: 293 K; reaction time: 3 h, except in entry 4 (10 h). Under the working conditions, the substrate (2 or 3) converted quantitatively or, if any, was detected in trace amounts. b Isolated yield (based on HetNCO2Ph) of HetNC(O)NRR′. | |||
| 1 | CH2CHCH2NH2 (0.96) |
2 (0.86) | 13: 92 |
| 2 | CH2CHCH2NH2 (0.96) |
3 (0.86) | 14: 64 |
| 3 | O(CH2CH2)2NH (0.80) | 2 (0.71) | 15: 92 |
| 4 | O(CH2CH2)2NH (0.80) | 3 (0.71) | 16: 61 |
 | ||
| Scheme 4 Aminolysis of HetNCO2Ph. | ||
Conclusion
The reaction of the N-phenoxycarbonyl derivatives of pyrrole, indole and carbazole, HetNCO2Ph, with amines has been shown to be a convenient method of synthesis of unsymmetrical ureas HetNC(O)NRR′ (HetNH = pyrrole, indole, carbazole). The aminolysis reaction can be catalyzed by the amidine base DBU under usually mild conditions and proceeded with good to excellent selectivity depending on the working conditions (substrate, amine, temperature). The protocol has been successfully extended also to functionalized amines, such as amino esters and amino alcohols.The synthetic approach is simple, direct, efficient and allows to gain access to the target products through a phosgene- and halogen-free synthetic pathway (Scheme 5; as a comparison, see also Scheme 1), which is safe and obviates the large co-generation of wastes (salts, etc.) typical of phosgenation methods. It provides a new solution for the synthesis of unsymmetrical ureas HetNC(O)NRR′ through a green route alternative to the current conventional procedures.
 | ||
| Scheme 5 Phosgeneless route to unsymmetrical ureas HetNC(O)NRR′. | ||
Experimental
General methods
All solvents were dried according to conventional methods (P2O5; Na/benzophenone)29 and stored under N2. The carbonyl derivatives 1–3 were obtained as previously reported.17dDBU and the amines were commercial products (Fluka or Aldrich) and were stored and manipulated under N2 to prevent any significant contamination by atmospheric CO2 and moisture. Ph-AE was purified by column chromatography (silica gel; MeOH as eluent) and recrystallized from EtOH(minimum amount)/diethyl ether/n-hexane before use. GC analyses were performed with a HP 5890 Series II gas-chromatograph (capillary column: Heliflex AT-5, 30 m × 0.25 mm, 0.25 μm film thickness). GC-MS analyses were carried out with a Shimadzu GC-17A linked to a Shimadzu GC-MS QP5050 selective mass detector (capillary column: Supelco MDN-5S, 30 m × 0.25 mm, 0.25 μm film thickness). IR spectra were taken on a Shimadzu FT-IR Prestige 21 spectrophotometer or recorded with a Perkin Elmer FT-IR 1710 instrument. Polarimetric measurements were carried out with a Perkin Elmer 343 Polarimeter. NMR spectra were run with a Varian Inova 400 spectrometer or a Bruker AM 500 instrument. Chemical shift are in δ (ppm) vs. Me4Si. Coupling constants are in Hz.Synthesis and characterization of unsymmetrical ureas HetNC(O)NRR′ (4–16)
The reactions were preferably conducted under N2 and carried out in a ≈20 mL glass reactor equipped with a Sovirel cap and a Thorion stopcock.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v)). Yield of 4: 194 mg, 90%. Found: C, 71.94; H, 6.09; N, 14.00. Calc. for C12H12N2O: C, 71.98; H, 6.04; N, 13.98%; νmax(Nujol)/cm−1 3321 s (NH), 1684vs (CO); δH (400 MHz; CDCl3; 293 K) 4.57 (2 H, d, J = 5.9, CH2), 5.78 (1 H, br, NH), 6.25 (2 H, t, J = 2.2, Hβ), 7.18 (2 H, t, J = 2.2, Hα), 7.26–7.38 (5 H, m, Ph); δC (100 MHz; CDCl3; 293 K) 44.91 (CH2), 111.99 (Cβ), 118.38 (Cα), 127.88 (Cortho), 127.91 (Cpara), 128.87 (Cmeta), 137.49 (Cipso), 150.87 (CO); m/z (EI) 200 (M+), 133 (M − C4H4NH), 104, 91, 67, 51, 39.
:1 v/v)). Yield of 5: 107 mg, 87%. Found: C, 64.10; H, 6.73; N, 18.59. Calc. for C8H10N2O: C, 63.99; H, 6.71; N, 18.65%; νmax(Nujol)/cm−1 3333 s (NH), 1682vs (CO); δH (500 MHz; CDCl3; 293 K) 4.00 (2 H, tt, 3JHCNH ≈ 3JHCCH = 5.8, 4J = 1.5, CH2), 5.18 (1 H, dq, 3Jcis = 10.3, 4J ≈ 2J = 1.4, Hcis), 5.24 (1 H, dq, 3Jtrans = 17.2, 4J ≈ 2J = 1.4, Htrans), 5.76 (1 H, br, NH), 5.89 (1 H, m, HCCH2), 6.25 (2 H, t, J = 2.3, Hβ), 7.19 (2 H, t, J = 2.3, Hα); δC (125 MHz; CDCl3; 293 K) 43.21 (CH2), 111.89 (Cβ), 117.05 (CH = CH2) 118.35 (Cα), 133.62 (HC = CH2),150.84 (CO); m/z (EI) 150 (M+), 135, 106, 94, 80, 67, 54, 41, 39.
:1 v/v)). Yield of 6: 180 mg, 93%. Found: C, 60.07; H, 6.80; N, 15.56. Calc. for C9H12N2O2: C, 59.99; H, 6.71; N, 15.54%; νmax(Nujol)/cm−1 1682vs (CO); δH (500 MHz; CDCl3; 293 K) 3.60 (4 H, t, J = 4.8, NCH2), 3.72 (4 H, t, OCH2), 6.22 (2 H, t, J = 2.2, Hβ), 6.98 (2 H, t, J = 2.3 Hz, Hα); δC (125 MHz; CDCl3; 293 K) 46.98 (NCH2), 66.56 (OCH2), 110.98 (Cβ), 120.33 (Cα), 153.88 (CO); m/z (EI) 180 (M+), 149, 135, 122, 114 (M − C4H4N), 94, 80, 70, 66, 56, 42.
O); δH (400 MHz; CDCl3; 293 K) 4.58 (4 H, s, CH2), 6.21 (2 H, t, J = 2.2, Hβ), 7.11 (2 H, t, J = 2.2, Hα), 7.23 (4 H, dm, Hortho), 7.31 (2 H, m, Hpara), 7.37 (4 H, m, Hmeta); δC (100 MHz; CDCl3; 293 K) 50.76 (CH2), 111.02 (Cβ), 120.59 (Cα), 127.73, 127.80, 128.88, 135.94 (Cipso) 155.14 (CO); m/z (EI) 290 (M+), 224 (M − C4H4N), 199 (M-PhCH2), 156, 132, 104, 91, 77, 65, 51, 39.
O); δH (400 MHz; CDCl3; 293 K) 6.31 (2 H, t, J = 2.2, Hβ), 7.15 (1 H, m, Hpara), 7.26 (1 H, br, NH), 7.28 (2 H, t, J = 2.2, Hα), 7.35 (2 H, m, Hmeta), 7.48 (2 H, dm, Hortho); δC (100 MHz; CDCl3; 293 K) 112.51 (Cβ), 118.53 (Cα), 120.41 (Cortho), 124.91, (Cpara), 129.22 (Cmeta), 136.76 (Cipso), 148.34 (CO); m/z (EI) 186 (M+), 119 (M − C4H4NH), 91, 67, 51, 39.
O); δH (400 MHz; CDCl3; 293 K) 3.44 (3 H, s, CH3), 5.97 (2 H, t, J = 2.2, Hβ), 6.74 (2 H, t, J = 2, Hα), 7.06 (2 H, dm, Hortho), 7.21 (1 H, m, Hpara), 7.31 (2 H, m, Hmeta); δC (100 MHz; CDCl3; 293 K) 40.11 (Me), 110.46 (Cβ), 121.14 (Cα), 125.45 (Cortho), 126.83 (Cpara), 129.81 (Cmeta), 144.48 (Cipso), 152.97 (CO); m/z (EI) 200 (M+), 134 (M − C4H4N), 119, 106, 77, 51, 39.
:1 and, then, 2:1 (v/v)). Yield of 10: 124 mg, 87%. The product can be recrystallized at 253 K from diethyl ether/n-hexane. Found: C, 60.61; H, 7.70; N, 11.68. Calc. for C12H18N2O3: C, 60.49; H, 7.61; N, 11.75%; νmax(Nujol)/cm−1 3325 s (NH), 1744vs (CO2Me), 1667vs (CO); δH (400 MHz; CDCl3; 293 K) 0.95 (3 H, d, 3J = 8, CH3), 0.97 (3 H, d, 3J = 8, CH3), 1.58–1.80 (3 H, overlapped multiplets, CH2 and CHMe2), 3.76 (3 H, s, OMe), 4.68 (1 H, td, 3JHCCH = 5, 3JHCCH ≈ 3JHCNH = 8, CHCO2Me), 5.79 (1 H, br d, 3JHCNH = 8, NH), 6.25 (2 H, t, J = 2, Hβ), 7.19 (2 H, t, J = 2, Hα); δC (100 MHz; CDCl3; 293 K) 21.91 (Me), 22.78 (Me), 24.83 (CHMe2), 41.79 (CH2), 51.99 (CHCO2Me), 52.57 (OMe), 112.11 (Cβ), 118.42 (Cα), 150.43 (CO), 173.38 (CO2Me); m/z (EI) 238 (M+), 179 (M − CO2Me), 163, 138, 112, 88, 67, 55, 41.
O), 1551vs; δH (400 MHz; CDCl3; 293 K) 2.67 (1 H, br s, OH), 3.52 (2 H, q, 3J = 5, NCH2), 3.77 (2 H, t, 3J = 5, CH2O), 6.23 (2 H, t, J = 2, Hβ), 6.26 (1 H, br, NH), 7.18 (2 H, t, J = 2.2, Hα); δC (100 MHz; CDCl3; 293 K) 43.12 (NCH2), 61.65 (CH2OH), 112.00 (Cβ), 118.39 (Cα), 151.73 (CO); m/z (EI) 154 (M+), 136 (M − H2O), 122 (M − MeOH), 106, 94, 80, 67, 56, 41.
:1 v/v) mixture as the eluent. Yield of 12: 118.5 mg, 80%.31 The product can be recrystallized (colorless plates) at 253 K from diethyl ether/n-hexane. Found: C, 67.90; H, 6.20; N, 12.09. Calc. for C13H14N2O2: C, 67.81; H, 6.13; N, 12.16%; νmax(Nujol)/cm−1 3294 s, 1674vs (CO), 1558 ms; δH (400 MHz; CDCl3; 293 K) 2.4 (1 H, very broad, OH), 3.42 (1 H, ddd, 2J = 13.9, 3JHCCH = 8.4, 3JHCNH = 4.4, CH2), 3.82 (1 H, ddd, 2J = 13.9, 3JHCCH = 3.5, 3JHCNH = 7.3, CH2), 4.92 (1 H, dd, 3JHCCH = 3.5, 3JHCCH = 8.3, CH), 5.99 (1 H, br, NH), 6.25 (2 H, t, J = 2, Hβ), 7.16 (2 H, t, J = 2, Hα), 7.26–7.40 (5 H, m, Ph); δC (100 MHz; CDCl3; 293 K) 48.02 (NCH2), 73.27 (CHOH), 112.05 (Cβ), 118.39 (Cα), 125.78, 128.24, 128.71, 141.20 (Cipso), 151.49 (CO); m/z (EI) 230 (M+), 213 (M − OH), 181, 156, 144, 124, 107, 94, 80, 67, 51, 39.
:1 v/v) mixture as the eluent. Yield of 13: 158.6 mg, 92%. Product 13 can be recrystallized at 253 K from diethyl ether(minimum amount)/n-hexane. Found: C, 71.90; H, 6.10; N, 13.90. Calc. for C12H12N2O: C, 71.98; H, 6.04; N, 13.98%; νmax(Nujol)/cm−1 3356 ms (NH), 1682vs and 1666vs (poorly resolved, CO), 1535vs; δH (400 MHz; CDCl3; 293 K) 4.09 (2 H, tt, 3JHCCH ≈ 3JHCNH = 5.5, 4J = 1.5, CH2), 5.21 (1 H, dq, Jcis = 10.2, 2J ≈ 4J = 1.5, Hcis), 5.30 (1 H, dq, Jtrans = 17.2, 2J ≈ 4J = 1.5 Hz, Htrans), 5.65 (1 H, br, NH), 5.96 (1 H, m, CH), 6.61 (1 H, dd, 3J = 3.7, 4J = 1.1, 3-H), 7.21 (1 H, m, 3J5,4 = 7.7, 3J5,6 = 7.3, 4J5,7 = 1.1, 5-H), 7.30 (1 H, m, 3J6,5 = 7.3, 3J6,7 = 8.1, 4J6,4 = 1.5, 6-H), 7.44 (1 H, d, 3J = 3.7, 2-H), 7.58 (1 H, dt, 3J4,5 = 7.7, 4J4,6 ≈ 4J4,3 = 1.5, 4-H), 8.06 (1 H, dm, 3J7,6 = 8, 7-H); δC (100 MHz; CDCl3; 293 K) 43.28 (CH2), 107.13, 114.01, 117.15, 121.22, 122.29, 123.96, 124.22, 130.17, 133.77, 135.03, 151.90 (CO); m/z (EI) 200 (M+), 130, 117, 89, 65, 63, 41, 39.
:1 v/v) mixture as the eluent. Yield of 14: 137.4 mg, 64%. The product can be recrystallized (colorless needles) at 253 K from diethyl ether(minimum amount)/n-hexane. Found: C, 76.85; H, 5.7; N, 11.09. Calc. for C16H14N2O: C, 76.78; H, 5.64; N, 11.19%; νmax(Nujol)/cm−1 3271 ms (NH), 1667vs (CO), 1528vs; δH (400 MHz; CDCl3; 293 K) 4.18 (2 H, tt, 3JHCCH ≈ 3JHCNH = 6, 4J = 1.5, CH2), 5.25 (1 H, dq, Jcis = 10.3, 2J ≈ 4J = 1.5 Hz, Hcis), 5.34 (1 H, dq, Jtrans = 17.2, 2J ≈ 4J = 1.5, Htrans), 5.77 (1 H, br, NH), 6.03 (1 H, m, CH), 7.33 (2 H, m), 7.47 (2 H, m), 8.02 (4 H, overlapped dm); δC (100 MHz; CDCl3; 293 K) 43.45 (CH2), 113.54, 117.43, 120.20, 122.29, 125.10, 126.95, 133.71, 138.27, 152.63 (CO); m/z (EI) 250 (M+), 167, 140, 115, 113, 83, 63, 56, 41.
:1 v/v) mixture as the eluent. The fractions containing the product were collected and the solvent was evaporated. The product, which was contaminated by minor amounts of O(CH2CH2)2NCO2Ph, was recrystallized at 253 K from diethyl ether/n-hexane. Yield of 15: 149.3 mg, 92%. Found: C, 67.90; H, 6.20; N, 12.10. Calc. for C13H14N2O2: C, 67.81; H, 6.13; N, 12.16%; νmax(Nujol)/cm−1 1682vs (CO); δH (400 MHz; CDCl3; 293 K) 3.59 (4 H, t, J = 4.7, NCH2), 3.77 (4 H, t, J = 5 Hz, OCH2), 6.60 (1 H, dd, 3J = 3.7, 4J = 1, 3-H), 7.19 (1 H, m, 3J5,4 = 7.7, 3J5,6 = 7.3, 4J5,7 = 1.1, 5-H), 7.29 (1 H, m, 3J6,5 = 7, 3J6,7 = 8.4, 4J6, 4 = 1.1, 6-H), 7.29 (1 H, d, 3J = 3.7, 2-H), 7.58 (1 H, dt, 3J4,5 = 7.7, 4J4, 6 ≈ 4J4, 3 = 1, H-4), 7.67 (1 H, dm, 3J7,6 = 8, 7-H); δC (100 MHz; CDCl3; 293 K) 47.05 (NCH2), 66.67 (OCH2), 106.27, 113.17, 121.12, 122.05, 123.75, 126.10, 129.59, 135.15, 154.27 (CO); m/z (EI) 230 (M+), 200, 144 (M − N(CH2CH2)2O), 130, 114 (M − C8H6N), 103, 89, 70, 63, 42.
:1 v/v) mixture as the eluent. The chromatographic separation allowed to recover pure carbazole and a mixture of O(CH2CH2)2NCO2Ph and the target urea, from which 16 was isolated (120.2 mg, 61%) by recrystallyzation at 253 K from diethyl ether with n-hexane. Found: C, 72.90; H, 5.80; N, 9.95. Calc. for C17H16N2O2: C, 72.84; H, 5.75; N, 9.99%; νmax(Nujol)/cm−1 1666vs (CO); δH (400 MHz; CDCl3; 293 K) 3.60 (4 H, t, J = 4.8, NCH2), 3.78 (4 H, t, J = 5, OCH2), 7.31 (2 H, m), 7.46 (2 H, m), 7.65 (2 H, dm, J = 8.1), 8.02 (2 H, dm, J = 7.7); δC (100 MHz; CDCl3; 293 K) 46.80 (NCH2), 66.84 (OCH2), 112.58, 120.24, 121.82, 124.48, 126.68, 138.44, 153.95 (CO); m/z (EI) 280 (M+), 194 (M − N(CH2CH2)2O), 180, 166 (M − OCN(CH2CH2)2O), 140, 114, 89, 70, 56, 42.
:1 v/v)). Yield of 4: 194 mg, 90%. Found: C, 71.94; H, 6.09; N, 14.00. Calc. for C12H12N2O: C, 71.98; H, 6.04; N, 13.98%; νmax(Nujol)/cm−1 3321 s (NH), 1684vs (CO); δH (400 MHz; CDCl3; 293 K) 4.57 (2 H, d, J = 5.9, CH2), 5.78 (1 H, br, NH), 6.25 (2 H, t, J = 2.2, Hβ), 7.18 (2 H, t, J = 2.2, Hα), 7.26–7.38 (5 H, m, Ph); δC (100 MHz; CDCl3; 293 K) 44.91 (CH2), 111.99 (Cβ), 118.38 (Cα), 127.88 (Cortho), 127.91 (Cpara), 128.87 (Cmeta), 137.49 (Cipso), 150.87 (CO); m/z (EI) 200 (M+), 133 (M − C4H4NH), 104, 91, 67, 51, 39.
:1 v/v)). Yield of 5: 107 mg, 87%. Found: C, 64.10; H, 6.73; N, 18.59. Calc. for C8H10N2O: C, 63.99; H, 6.71; N, 18.65%; νmax(Nujol)/cm−1 3333 s (NH), 1682vs (CO); δH (500 MHz; CDCl3; 293 K) 4.00 (2 H, tt, 3JHCNH ≈ 3JHCCH = 5.8, 4J = 1.5, CH2), 5.18 (1 H, dq, 3Jcis = 10.3, 4J ≈ 2J = 1.4, Hcis), 5.24 (1 H, dq, 3Jtrans = 17.2, 4J ≈ 2J = 1.4, Htrans), 5.76 (1 H, br, NH), 5.89 (1 H, m, HCCH2), 6.25 (2 H, t, J = 2.3, Hβ), 7.19 (2 H, t, J = 2.3, Hα); δC (125 MHz; CDCl3; 293 K) 43.21 (CH2), 111.89 (Cβ), 117.05 (CH = CH2) 118.35 (Cα), 133.62 (HC = CH2),150.84 (CO); m/z (EI) 150 (M+), 135, 106, 94, 80, 67, 54, 41, 39.
:1 v/v)). Yield of 6: 180 mg, 93%. Found: C, 60.07; H, 6.80; N, 15.56. Calc. for C9H12N2O2: C, 59.99; H, 6.71; N, 15.54%; νmax(Nujol)/cm−1 1682vs (CO); δH (500 MHz; CDCl3; 293 K) 3.60 (4 H, t, J = 4.8, NCH2), 3.72 (4 H, t, OCH2), 6.22 (2 H, t, J = 2.2, Hβ), 6.98 (2 H, t, J = 2.3 Hz, Hα); δC (125 MHz; CDCl3; 293 K) 46.98 (NCH2), 66.56 (OCH2), 110.98 (Cβ), 120.33 (Cα), 153.88 (CO); m/z (EI) 180 (M+), 149, 135, 122, 114 (M − C4H4N), 94, 80, 70, 66, 56, 42.
O); δH (400 MHz; CDCl3; 293 K) 4.58 (4 H, s, CH2), 6.21 (2 H, t, J = 2.2, Hβ), 7.11 (2 H, t, J = 2.2, Hα), 7.23 (4 H, dm, Hortho), 7.31 (2 H, m, Hpara), 7.37 (4 H, m, Hmeta); δC (100 MHz; CDCl3; 293 K) 50.76 (CH2), 111.02 (Cβ), 120.59 (Cα), 127.73, 127.80, 128.88, 135.94 (Cipso) 155.14 (CO); m/z (EI) 290 (M+), 224 (M − C4H4N), 199 (M-PhCH2), 156, 132, 104, 91, 77, 65, 51, 39.
O); δH (400 MHz; CDCl3; 293 K) 6.31 (2 H, t, J = 2.2, Hβ), 7.15 (1 H, m, Hpara), 7.26 (1 H, br, NH), 7.28 (2 H, t, J = 2.2, Hα), 7.35 (2 H, m, Hmeta), 7.48 (2 H, dm, Hortho); δC (100 MHz; CDCl3; 293 K) 112.51 (Cβ), 118.53 (Cα), 120.41 (Cortho), 124.91, (Cpara), 129.22 (Cmeta), 136.76 (Cipso), 148.34 (CO); m/z (EI) 186 (M+), 119 (M − C4H4NH), 91, 67, 51, 39.
O); δH (400 MHz; CDCl3; 293 K) 3.44 (3 H, s, CH3), 5.97 (2 H, t, J = 2.2, Hβ), 6.74 (2 H, t, J = 2, Hα), 7.06 (2 H, dm, Hortho), 7.21 (1 H, m, Hpara), 7.31 (2 H, m, Hmeta); δC (100 MHz; CDCl3; 293 K) 40.11 (Me), 110.46 (Cβ), 121.14 (Cα), 125.45 (Cortho), 126.83 (Cpara), 129.81 (Cmeta), 144.48 (Cipso), 152.97 (CO); m/z (EI) 200 (M+), 134 (M − C4H4N), 119, 106, 77, 51, 39.
:1 and, then, 2:1 (v/v)). Yield of 10: 124 mg, 87%. The product can be recrystallized at 253 K from diethyl ether/n-hexane. Found: C, 60.61; H, 7.70; N, 11.68. Calc. for C12H18N2O3: C, 60.49; H, 7.61; N, 11.75%; νmax(Nujol)/cm−1 3325 s (NH), 1744vs (CO2Me), 1667vs (CO); δH (400 MHz; CDCl3; 293 K) 0.95 (3 H, d, 3J = 8, CH3), 0.97 (3 H, d, 3J = 8, CH3), 1.58–1.80 (3 H, overlapped multiplets, CH2 and CHMe2), 3.76 (3 H, s, OMe), 4.68 (1 H, td, 3JHCCH = 5, 3JHCCH ≈ 3JHCNH = 8, CHCO2Me), 5.79 (1 H, br d, 3JHCNH = 8, NH), 6.25 (2 H, t, J = 2, Hβ), 7.19 (2 H, t, J = 2, Hα); δC (100 MHz; CDCl3; 293 K) 21.91 (Me), 22.78 (Me), 24.83 (CHMe2), 41.79 (CH2), 51.99 (CHCO2Me), 52.57 (OMe), 112.11 (Cβ), 118.42 (Cα), 150.43 (CO), 173.38 (CO2Me); m/z (EI) 238 (M+), 179 (M − CO2Me), 163, 138, 112, 88, 67, 55, 41.
O), 1551vs; δH (400 MHz; CDCl3; 293 K) 2.67 (1 H, br s, OH), 3.52 (2 H, q, 3J = 5, NCH2), 3.77 (2 H, t, 3J = 5, CH2O), 6.23 (2 H, t, J = 2, Hβ), 6.26 (1 H, br, NH), 7.18 (2 H, t, J = 2.2, Hα); δC (100 MHz; CDCl3; 293 K) 43.12 (NCH2), 61.65 (CH2OH), 112.00 (Cβ), 118.39 (Cα), 151.73 (CO); m/z (EI) 154 (M+), 136 (M − H2O), 122 (M − MeOH), 106, 94, 80, 67, 56, 41.
:1 v/v) mixture as the eluent. Yield of 12: 118.5 mg, 80%.31 The product can be recrystallized (colorless plates) at 253 K from diethyl ether/n-hexane. Found: C, 67.90; H, 6.20; N, 12.09. Calc. for C13H14N2O2: C, 67.81; H, 6.13; N, 12.16%; νmax(Nujol)/cm−1 3294 s, 1674vs (CO), 1558 ms; δH (400 MHz; CDCl3; 293 K) 2.4 (1 H, very broad, OH), 3.42 (1 H, ddd, 2J = 13.9, 3JHCCH = 8.4, 3JHCNH = 4.4, CH2), 3.82 (1 H, ddd, 2J = 13.9, 3JHCCH = 3.5, 3JHCNH = 7.3, CH2), 4.92 (1 H, dd, 3JHCCH = 3.5, 3JHCCH = 8.3, CH), 5.99 (1 H, br, NH), 6.25 (2 H, t, J = 2, Hβ), 7.16 (2 H, t, J = 2, Hα), 7.26–7.40 (5 H, m, Ph); δC (100 MHz; CDCl3; 293 K) 48.02 (NCH2), 73.27 (CHOH), 112.05 (Cβ), 118.39 (Cα), 125.78, 128.24, 128.71, 141.20 (Cipso), 151.49 (CO); m/z (EI) 230 (M+), 213 (M − OH), 181, 156, 144, 124, 107, 94, 80, 67, 51, 39.
:1 v/v) mixture as the eluent. Yield of 13: 158.6 mg, 92%. Product 13 can be recrystallized at 253 K from diethyl ether(minimum amount)/n-hexane. Found: C, 71.90; H, 6.10; N, 13.90. Calc. for C12H12N2O: C, 71.98; H, 6.04; N, 13.98%; νmax(Nujol)/cm−1 3356 ms (NH), 1682vs and 1666vs (poorly resolved, CO), 1535vs; δH (400 MHz; CDCl3; 293 K) 4.09 (2 H, tt, 3JHCCH ≈ 3JHCNH = 5.5, 4J = 1.5, CH2), 5.21 (1 H, dq, Jcis = 10.2, 2J ≈ 4J = 1.5, Hcis), 5.30 (1 H, dq, Jtrans = 17.2, 2J ≈ 4J = 1.5 Hz, Htrans), 5.65 (1 H, br, NH), 5.96 (1 H, m, CH), 6.61 (1 H, dd, 3J = 3.7, 4J = 1.1, 3-H), 7.21 (1 H, m, 3J5,4 = 7.7, 3J5,6 = 7.3, 4J5,7 = 1.1, 5-H), 7.30 (1 H, m, 3J6,5 = 7.3, 3J6,7 = 8.1, 4J6,4 = 1.5, 6-H), 7.44 (1 H, d, 3J = 3.7, 2-H), 7.58 (1 H, dt, 3J4,5 = 7.7, 4J4,6 ≈ 4J4,3 = 1.5, 4-H), 8.06 (1 H, dm, 3J7,6 = 8, 7-H); δC (100 MHz; CDCl3; 293 K) 43.28 (CH2), 107.13, 114.01, 117.15, 121.22, 122.29, 123.96, 124.22, 130.17, 133.77, 135.03, 151.90 (CO); m/z (EI) 200 (M+), 130, 117, 89, 65, 63, 41, 39.
:1 v/v) mixture as the eluent. Yield of 14: 137.4 mg, 64%. The product can be recrystallized (colorless needles) at 253 K from diethyl ether(minimum amount)/n-hexane. Found: C, 76.85; H, 5.7; N, 11.09. Calc. for C16H14N2O: C, 76.78; H, 5.64; N, 11.19%; νmax(Nujol)/cm−1 3271 ms (NH), 1667vs (CO), 1528vs; δH (400 MHz; CDCl3; 293 K) 4.18 (2 H, tt, 3JHCCH ≈ 3JHCNH = 6, 4J = 1.5, CH2), 5.25 (1 H, dq, Jcis = 10.3, 2J ≈ 4J = 1.5 Hz, Hcis), 5.34 (1 H, dq, Jtrans = 17.2, 2J ≈ 4J = 1.5, Htrans), 5.77 (1 H, br, NH), 6.03 (1 H, m, CH), 7.33 (2 H, m), 7.47 (2 H, m), 8.02 (4 H, overlapped dm); δC (100 MHz; CDCl3; 293 K) 43.45 (CH2), 113.54, 117.43, 120.20, 122.29, 125.10, 126.95, 133.71, 138.27, 152.63 (CO); m/z (EI) 250 (M+), 167, 140, 115, 113, 83, 63, 56, 41.
:1 v/v) mixture as the eluent. The fractions containing the product were collected and the solvent was evaporated. The product, which was contaminated by minor amounts of O(CH2CH2)2NCO2Ph, was recrystallized at 253 K from diethyl ether/n-hexane. Yield of 15: 149.3 mg, 92%. Found: C, 67.90; H, 6.20; N, 12.10. Calc. for C13H14N2O2: C, 67.81; H, 6.13; N, 12.16%; νmax(Nujol)/cm−1 1682vs (CO); δH (400 MHz; CDCl3; 293 K) 3.59 (4 H, t, J = 4.7, NCH2), 3.77 (4 H, t, J = 5 Hz, OCH2), 6.60 (1 H, dd, 3J = 3.7, 4J = 1, 3-H), 7.19 (1 H, m, 3J5,4 = 7.7, 3J5,6 = 7.3, 4J5,7 = 1.1, 5-H), 7.29 (1 H, m, 3J6,5 = 7, 3J6,7 = 8.4, 4J6, 4 = 1.1, 6-H), 7.29 (1 H, d, 3J = 3.7, 2-H), 7.58 (1 H, dt, 3J4,5 = 7.7, 4J4, 6 ≈ 4J4, 3 = 1, H-4), 7.67 (1 H, dm, 3J7,6 = 8, 7-H); δC (100 MHz; CDCl3; 293 K) 47.05 (NCH2), 66.67 (OCH2), 106.27, 113.17, 121.12, 122.05, 123.75, 126.10, 129.59, 135.15, 154.27 (CO); m/z (EI) 230 (M+), 200, 144 (M − N(CH2CH2)2O), 130, 114 (M − C8H6N), 103, 89, 70, 63, 42.
:1 v/v) mixture as the eluent. The chromatographic separation allowed to recover pure carbazole and a mixture of O(CH2CH2)2NCO2Ph and the target urea, from which 16 was isolated (120.2 mg, 61%) by recrystallyzation at 253 K from diethyl ether with n-hexane. Found: C, 72.90; H, 5.80; N, 9.95. Calc. for C17H16N2O2: C, 72.84; H, 5.75; N, 9.99%; νmax(Nujol)/cm−1 1666vs (CO); δH (400 MHz; CDCl3; 293 K) 3.60 (4 H, t, J = 4.8, NCH2), 3.78 (4 H, t, J = 5, OCH2), 7.31 (2 H, m), 7.46 (2 H, m), 7.65 (2 H, dm, J = 8.1), 8.02 (2 H, dm, J = 7.7); δC (100 MHz; CDCl3; 293 K) 46.80 (NCH2), 66.84 (OCH2), 112.58, 120.24, 121.82, 124.48, 126.68, 138.44, 153.95 (CO); m/z (EI) 280 (M+), 194 (M − N(CH2CH2)2O), 180, 166 (M − OCN(CH2CH2)2O), 140, 114, 89, 70, 56, 42.
Acknowledgements
Università degli Studi di Bari (Fondi di Ateneo) and Ministero dell'Istruzione, dell'Università e della Ricerca (PRIN 2008A7P7YJ_002) are acknowledged for financial support. The Authors thank Dr C. Cardellicchio (ICCOM-CNR, Bari) for polarimetric analyses.Notes and references
- P. T. Anastas and S. C. Warner, Green Chemistry. Theory and Practice, Oxford University Press, New York, 1998 Search PubMed.
- I. Gallou, Org. Prep. Proced. Int., 2007, 39, 355–383 CrossRef CAS; I Gallou, M. Eriksson, X. Zeng, C. Senanayake and V. Farina, J. Org. Chem., 2005, 70, 6960–6963 CrossRef CAS.
- F. Bigi, R. Maggi and G. Sartori, Green Chem., 2000, 2, 140–148 RSC.
- P. Hanson and D. A. R. Williams, J. Chem. Soc., Perkin Trans. 2, 1973, 2162–2165 RSC.
- L. L. Chapoy, D. Biddle, J. Halstrom, K. Kovacs, K. Brunfeldt, M. A. Qasim and T. Christensen, Macromolecules, 1983, 16, 181–186 Search PubMed.
- (a) M. Alvarez, D. Fernandez and J. A. Joule, J. Chem. Soc., Perkin Trans. 1, 1999, 249–255 RSC; (b) S. H. Hosseini and A. Entezami, Eur. Polym. J., 1995, 31, 635–641 Search PubMed; (c) D. J. Schipper, M. Hutchinson and K. Fagnou, J. Am. Chem. Soc., 2010, 132, 6910–6911 CrossRef CAS.
- J.-M. Gaudin, WO 016583, 2009; S. Allenmark, Tetrahedron Lett., 1990, 31, 1455–1458 Search PubMed.
- A. W. Weston, R. W. DeNet and R. J. Michaelis Jr, J. Am. Chem. Soc., 1953, 75, 4006–4008 Search PubMed; H. Takayama, Y. Yaegashi, M. Kitajima, X. Han, K. Nishimura, S. Okuyama and K. Igarashi, Bioorg. Med. Chem. Lett., 2007, 17, 4729–4732 Search PubMed; A. M. Felix, L. B. Czyzewski, D. P. Winter and R. I. Fryer, J. Med. Chem., 1969, 12, 384–387 CrossRef CAS.
- P. Cao, X.-F. Huang, H. Ding, H.-M. Ge, H.-Q. Li, B.-F. Ruan and H.-L. Zhu, Chem. Biodiversity, 2007, 4, 881–885 Search PubMed.
- (a) E. P. Papadopulos and H. S. Harry, J. Org. Chem., 1966, 31, 327–329 Search PubMed; (b) E. P. Papadopulos and H. S. Harry, J. Org. Chem., 1968, 33, 4551–4554 Search PubMed; (c) D. Ye, J. Wang, X. Zhang, Y. Zhou, X. Ding, E. Feng, H. Sun, G. Liu, H. Jiang and H. Liu, Green Chem., 2009, 11, 1201–1208 RSC; (d) W.-H. Fan, M. Parikh and J. K. Snyder, Tetrahedron Lett., 1995, 36, 6591–6594 CrossRef CAS; (e) J. E. Macor, A. Cuff and L. Cornelius, Tetrahedron Lett., 1999, 40, 2733–2736 CrossRef CAS; (f) A. Gimeno, M. Medio-Simon, C. Ramirez de Arellano, G. Asensio and A. B. Cuenca, Org. Lett., 2010, 12, 1900–1903 CrossRef CAS.
- L. Cotarca and H. Eckert, Phosgenations - A Handbook, Wiley-VCH, Weinheim, 2004, and references therein Search PubMed; J. S. Gift, R. McGaughy, D. V. Singh and B. Sonawane, Regul. Toxicol. Pharmacol., 2008, 51, 98–107 Search PubMed; S. A. Cucinell, Arch. Environ. Health, 1974, 28, 272–274 Search PubMed; W. F. Diller, Toxicol. Ind. Health, 1985, 1, 7–15 Search PubMed.
- For instance, the reaction of phenylisocyanate with pyrryl potassium afforded selectively 1-phenylaminocarbonyl pyrrole, while the analogous reaction with pyrryl magnesium bromide gave a mixture of 1- and 2-phenylaminocarbonyl pirrole.10a.
- (a) D. L. Boger and M. Patel, J. Org. Chem., 1987, 52, 2319–2323 Search PubMed; (b) D. L. Boger and M. Patel, J. Org. Chem., 1987, 52, 3934–3936 CrossRef CAS.
- Via ethylisocyanate: N. Goyal, Synlett, 2010, 335–336 Search PubMed.
- (a) M. Aresta and E. Quaranta, ChemTech, 1997, 27(3), 32–40 CAS; (b) E. Quaranta and M. Aresta, The Chemistry of N–CO2 Bonds: Synthesis of Carbamic Acids and Their Derivatives, Isocyanates, and Ureas, in Carbon Dioxide as Chemical Feedstock (ed. M. Aresta), Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2010, pp. 121–167 Search PubMed; (c) D. Ballivet-Tkatchenko and A. Dibenedetto, Synthesis of Linear and Cyclic Carbonates, in Carbon Dioxide as Chemical Feedstock (ed. M. Aresta), Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2010, pp. 169–212 Search PubMed.
- M. Carafa and E. Quaranta, Mini-Rev. Org. Chem., 2009, 6, 168–183 Search PubMed , and references therein.
- (a) M. Carafa, E. Mesto and E. Quaranta, Eur. J. Org. Chem., 2011, 2458–2465 Search PubMed; (b) E. Quaranta, M. Carafa and F. Trani, Appl. Catal., B, 2009, 91, 380–388 Search PubMed; (c) M. Distaso and E. Quaranta, J. Catal., 2008, 253, 278–288 Search PubMed; (d) M. Carafa, M. Distaso, V. Mele, F. Trani and E. Quaranta, Tetrahedron Lett., 2008, 49, 3691–3696 Search PubMed; (e) M. Distaso and E. Quaranta, Appl. Catal., B, 2006, 66, 72–80 CrossRef CAS; (f) M. Distaso and E. Quaranta, J. Catal., 2004, 228, 36–42 CrossRef CAS; (g) M. Distaso and E. Quaranta, Tetrahedron, 2004, 60, 1531–1539 CrossRef CAS.
- S. Fan, N. Zhao, J. Li, F. Xiao, W. Wei and Y. Sun, Catal. Lett., 2007, 120, 299–302 Search PubMed; X. Fu, Z. Zhang, C. Li, L. Wang, H. Ji, Y. Yang, T. Zou and G. Gao, Catal. Commun., 2009, 10, 665–668 Search PubMed; T. Baba, M. Fujiwara, A. Oosaku, A. Kobayashi, R. G. Deleon and Y. Ono, Appl. Catal., A, 2002, 227, 1–6 CrossRef CAS; T. Sima, S. Guo, F. Shi and Y. Deng, Tetrahedron Lett., 2002, 43, 8145–8147 CrossRef CAS; I. Vauthey, F. Valot, C. Gozzi, F. Fache and M. Lemaire, Tetrahedron Lett., 2000, 41, 6347–6350 CrossRef CAS; M. Curini, F. Epifano, F. Maltese and O. Rosati, Tetrahedron Lett., 2002, 43, 4895–4897 CrossRef CAS; M. Selva, P. Tundo, A. Perosa and F. Dall'Acqua, J. Org. Chem., 2005, 70, 2771–2777 CrossRef CAS; P. Tundo, L. Rossi and A. Loris, J. Org. Chem., 2005, 70, 2219–2224 CrossRef CAS; S. Ouk, S. Thiébaud, E. Borredon and B. Chabaud, Synth. Commun., 2005, 35, 3021–3026 CrossRef CAS; S. Ouk, S. Thiébaud, E. Borredon and P. Le Gars, Appl. Catal., A, 2003, 241, 227–233 CrossRef CAS; R. Juarez, A. Padilla, A. Corma and H. Garela, Ind. Eng. Chem. Res., 2008, 47, 8043–8047 Search PubMed; R. Juarez, H. Pennemann and H. Garcia, Catal. Today, 2011, 159, 25–28 Search PubMed.
- D. Delledonne, F. Rivetti and U. Romano, Appl. Catal., A, 2001, 221, 241–251 CrossRef; T. Matsuzaki and A. Nakamura, Catal. Surv. Jpn., 1997, 1, 77–88 CrossRef CAS; S. Cshihony, L. T. Mika, G. Vlad, K. Barta, C. P. Mehnert and I. T. Horvath, Collect. Czech. Chem. Commun., 2007, 72, 1094 CrossRef CAS.
- B. M. Bhanage, S.-I. Fujita, Y. Ikushima and M. Arai, Appl. Catal., A, 2001, 219, 259–266 CrossRef CAS.
- L. Grehn and U. Ragnarsson, Angew. Chem., Int. Ed. Engl., 1984, 23, 296–301 CrossRef; L. A. Carpino and D. E. Barr, J. Org. Chem., 1966, 31, 764–767 CrossRef CAS; J. Bergman, R. Carlsson and B. Sjoberg, J. Heterocycl. Chem., 1977, 14, 1123–1133 CAS; N. C. Wang and H. J. Anderson, Can. J. Chem., 1977, 55, 4103–4111 Search PubMed; N. Gabel, J. Org. Chem., 1962, 27, 301–303 CAS; R. M. Acheson and J. M. Vernon, J. Chem. Soc., Chem. Commun., 1961, 457–459 Search PubMed; S. T. Handy, J. J. Sabatini, Y. Zhang and I. Vulfova, Tetrahedron Lett., 2004, 45, 5057–5060 CrossRef CAS; U. Jacquemard, V. Bénéteau, M. Lefoix, S. Routier, J.-Y. Mérour and G. Coudert, Tetrahedron, 2004, 60, 10039–10047 CrossRef CAS; V. O. Illi, Synthesis, 1979, 387–388 Search PubMed; T. Itahara, Heterocycles, 1986, 24, 2557–2562 Search PubMed.
- Both diethyl ether and THF were inert towards the reactants employed in this study and solubilized most of them very well. We have deliberately avoided the use of halogenated and aromatic solvents because of their very unfavorable ecological impact. The use of other common solvents, such as H2O, alcohols, acetone, was prevented by the poor substrate solubility and/or the chemical behavior of the reactants in these media.
- A. S. Shawaly, A. Harbash, M. M. Sidky, H. M. Hassaneen and S. S. Elkaabi, J. Org. Chem., 1986, 51, 3498–3501 CrossRef CAS; K. Sung, B.-R.- Zhuang, P.-M.- Huang and S. W. Jhong, J. Org. Chem., 2008, 73, 4027–4033 Search PubMed.
- GC-MS of (PhCH2)2NCO2Ph: m/z (EI) 317 (M+), 224 (M − OPh), 210, 132, 91, 77, 65, 39.
- From comparing the pKa of DBUH+ (13.9)26 in DMSO with that of H3N+CH2CO2Et (8.7)27 in the same solvent (DMSO), it can be reasonably inferred that DBU is a stronger base than the L-leucine methyl ester.
- R. Schwesinger, H. Schlemper, C. Hasenfratz, J. Willaredt, T. Dambacher, T. Breuer, C. Ottaway, M. Fletschinger, J. Boele, H. Fritz, D. Putzas, H. W. Rotter, F. G. Bordwell, A. V. Satish, G.-Z. Ji, E. M. Peters, H. G. Von Schnering and L. Walz, Liebigs Ann., 1996, 1055–1081 Search PubMed.
- F. G. Bordwell, Acc. Chem. Res., 1988, 21, 456–463 CrossRef CAS.
- Polarimetric measurements on isolated urea10 have shown that the ureidization process was accompanied by extended racemization of the chiral center of the amino ester moiety.
- D. D. Perrin, W. L. F. Armarego and D. R. Perrin, Purification of Laboratory Chemicals, Pergamon, Oxford, 1986 Search PubMed.
- From the chromatographic separation we isolated an impure fraction mainly containing the side-product 17 characterized as C4H4NC(O)NHCH2CH2O(O)CNC4H4 on the basis of the NMR and mass spectra. δH (400 MHz; CDCl3; 293 K) 3.77 (2 H, q, J = 5, NCH2), 4.54 (2 H, t, J = 5, CH2O), 6.24 (1 H, br unresolved t, NH), 6.25 (2 H, t, J = 2, Hβ), 6.26 (2 H, t, J = 2, Hβ), 7.18 (2 H, t, J = 2, Hα,ureidic), 7.25 (2 H, t, J = 2, Hα,carbamic); δC (100 MHz; CDCl3; 293 K) 40.52 (NCH2), 65.79 (CH2O), 112.26 (Cβ,ureidic), 113.05 (Cβ,carbamic), 118.39 (Cα,ureidic), 120.09 (Cα,carbamic), 150.89 (C(O)O), 151.19 (CO); m/z (EI) 247 (M+), 180 (M − C4H5N), 137, 111, 94, 80, 67, 42.
- From the chromatographic separation we collected an impure fraction mainly containing the side-product 18 characterized as C4H4NC(O)NHCH2CH(Ph)O(O)CNC4H4 on the basis of the NMR and mass spectra. δH (400 MHz; CDCl3; 293 K) 3.90–3.96 (2 H, m, NCH2), 5.95 (1 H, br unresolved t, NH), 6.07 (1 H, dd, J = 4, 8, OCH), 6.24 (2 H, t, J = 2 Hβ,ureidic), 6.25 (2 H, t, J = 2, Hβ,carbamic), 7.13 (2 H, t, J = 2, Hα,ureidic), 7.28 (2 H, t, J = 2, Hα,carbamic), 7.34–7.46 (5 H, m, Ph); δC (100 MHz; CDCl3; 293 K) 45.98 (NCH2), 77.88 (CH), 112.15 (Cβ,ureidic), 113.01 (Cβ,carbamic), 118.38 (Cα,ureidic), 120.15 (Cα,carbamic),126.36, 129.05, 129.18, 136.36 (Cipso), 150.10 (C(O)O), 150.87 (CO); m/z (EI) 323 (M+), 281, 256, 213, 184, 170, 156, 146, 128, 104, 94, 77, 68, 51, 39.
| This journal is © The Royal Society of Chemistry 2012 |