An exceptionally active and selective Pt–Au/TiO2 catalyst for hydrogenation of the nitro group in chloronitrobenzene†
Daiping
He
*a,
Xiangdong
Jiao
a,
Ping
Jiang
a,
Jian
Wang
a and
Bo-Qing
Xu
b
aKey Lab of Advanced Materials, College of Chemistry, Chongqing Normal University, Chongqing, 401331, China. E-mail: hedaiping@126.com; Fax: (+86)23-6536-2777; Tel: (+86)23-6536-2777
bInnovative Catalysis Program, Key Lab of Organic Optoelectronics & Molecular Engineering, Department of Chemistry, Tsinghua University, Beijing, 100084, China. E-mail: bqxu@mail.tsinghua.edu.cn; Fax: (+86)10-6279-2122; Tel: (+86)10-6277-2592
First published on 28th October 2011
Abstract
Adding a very small amount of Pt entities (0.01–0.03 wt%) onto the Au surface of a Au/TiO2 catalyst is found to be an efficient approach to improve the catalytic activity of Au for the hydrogenation of p-chloronitrobenzene (p-CNB), without loss of selectivity towards p-chloroaniline (p-CAN). The effect of catalyst amount, reaction temperature, H2 pressure and reaction time on p-CNB hydrogenation was studied with 0.02wt%Pt–0.5wt%Au/TiO2 (Pt0.0002–Au0.005/TiO2). The selectivity to p-CAN could be up to 100% at complete conversion of p-CNB with reaction temperatures at or below 333 K. The catalyst also exhibited perfect stability. The catalyst structure was characterized by TEM and XRD, and the mechanism of the high activity of the catalyst was discussed.
1. Introduction
Chloroanilines are an important class of industrial intermediates for a variety of specific and fine chemicals, including pharmaceuticals, dyes, herbicides and pesticides.1p-CAN, a high production volume compound, can be synthesised viaFe promoted reduction of p-CNB in acid media (Bechamp reaction) or by selective hydrogenation using transition-metal catalysts. Industrial implementation of the Bechamp reaction is no longer viable due to the generation of significant amounts of toxic metal waste which requires costly and laborious separation and disposal.2 Liquid phase catalytic hydrogenation has emerged as a cleaner alternative to the Bechamp process with higher associated product yields.3 However, the economic benefits are dependent on catalyst performance, where unwanted C–Cl bond hydrogenolysis is difficult to fully avoid. Reaction selectivity is critical as p-CNB hydrogenation can generate a range of intermediates and by-products as shown in Scheme 1, which presents the reaction pathways proposed for batch liquid phase hydrogenation.4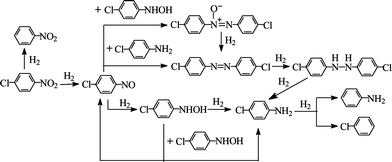 | ||
| Scheme 1 The hydrogenation reaction pathways of p-CNB. | ||
With a variety of catalysts (e.g., platinum, palladium, rhodium, nickel), the hydrogenation of p-CNB to the corresponding p-CAN is always accompanied by some dechlorination reaction. Furthermore, hydrogen chloride (HCl) produced from the dechlorination contributes to the corrosion of the reactor. To achieve high selectivity towards p-CAN, great attempts have been made to tailor the hydrogenation selectivity of supported metal catalysts toward a specific hydrogenation of the nitro group in p-CNB, e.g., by modifying the active metal with different promoters5 or a second metal component,6 by assembling metal-colloids onto suitable oxide nanoparticles7 or using amorphous structures8 and by using delicate approaches for catalyst preparation.9 However, the side reaction of hydrodechlorination still cannot be avoided completely.
Having started a couple of decades ago, catalysis by gold continues to show a fascinating future in the field of catalysis.10 Nanosized Au particles in interaction with a variety of support materials showed special selectivity in catalyzing a number of reactions including CO oxidation,11alcohol oxidation,12olefin epoxidation,13vinyl chloride synthesis,14 selective hydrogenation of unsaturated hydrocarbons,15aldehydes and ketones.16 For the reduction of nitro compounds with H2, Au/TiO2, Au/Fe2O3 and Au/SiO2 catalysts were recently found to be very specific in reducing the nitro group in various compounds containing other functional groups.17 Our previous work also found that selective production of chloroanilines from chloronitrobenzenes without any dechlorination was feasible by catalytic hydrogenation over Au/ZrO2.18 However, the hydrogenation activity of Au nanoparticles was often much lower than those of the platinum-group metals,19 most probably due to an “intrinsic” nobleness of Au for H2activation, that the dissociative adsorption of H2 on Au is an activated process and is restricted to the edge and corner sites or at the interface perimeter around the Au nanoparticles in contact with oxide support.20 Such low hydrogenation activity of the Au nanoparticles has been a major obstacle in view of practical applications.
In the present study, we report that adding a very small amount of Pt entities (0.01–0.03 wt%) onto the Au surface of a Au/TiO2 catalyst is an efficient approach to improve the catalytic activity of Au for the hydrogenation of p-chloronitrobenzene, without changing the selectivity propensity of the Au nanoparticles for the reaction. Our results can provide the basis for the development of a clean, high efficient route for the production of p-CAN under mild conditions.
2. Results and discussion
Table 1 shows the catalytic results of p-CNB hydrogenation over a Pt–Au/TiO2 catalyst at 323 K, 1.0 MPa H2. Pt/TiO2 and Au/TiO2 catalysts were also included for comparison. Blank tests showed no reaction of p-CNB in the absence of the catalyst. The conversion of p-CNB over the 0.01 wt%Pt/TiO2 (Pt0.0001/TiO2) was not detected at all. The 0.5 wt%Au/TiO2 (Au0.005/TiO2) produced a low p-CNB conversion (only 0.9%) in 1 h. When the same Au0.005/TiO2 were loaded with a very small amount of Pt (only 0.01 wt%), however, a dramatically higher p-CNB conversion (99.2%) was achieved under the same reaction conditions, and the undesired hydrodechlorination reaction usually observed over other metal catalysts21 was not detected, which indicated that the C–Cl bond in the p-CNB molecule was kept intact during the reaction. In addition, the selectivity for p-CAN increased from 94.7% to 99.9%, most probably due to the intermediates22 (4, 4′-dichloroazoxybenzene and 4, 4′-dichloroazobenzene) produced in the reaction process being further hydrogenated to the desired product p-CAN, which was supported by the observed increase in the selectivity of the desired p-CAN with increasing p-CNB conversion. Increasing the amount of Pt up to 0.03 wt% in the Pt–Au0.005/TiO2 catalyst resulted in continued shortening of the reaction period for achieving a complete conversion of p-CNB. For instance, a reaction period of 40 min was found to be long enough to produce a p-CNB conversion of 100% over the Pt0.0003–Au0.005/TiO2 catalyst. With the amount of Pt in the Pt–Au0.005/TiO2 increasing continuously, the reaction period for achieving a complete conversion of p-CNB was further reduced, but the selectivity of p-CAN began to decrease due to hydrodechlorination, and so the selectivity of aniline (AN) increased. For example, the selectivities for p-CAN and AN were 99.9% and 0.1% over Pt0.0004–Au0.005/TiO2, respectively. Galvagno et al.23 reported that the activity of bimetallic M–Pt is enhanced at low M/Pt ratios, whereas at high M content the platinum is poisoned. With the amount of Pt in the Pt–Au0.005/TiO2 increasing continuously, the Au/Pt ratio reduced, so that the activity of Pt–Au0.005/TiO2 was probably enhanced to result in hydrodechlorination. Therefore, the amount of Pt in the Pt–Au0.005/TiO2 may be restricted to 0.03 wt%.| Sel. (%) | |||||
|---|---|---|---|---|---|
| Catalyst | React. time (min) | p-CNB Conv. (%) | p-CAN | AN | Othersb |
| n.d.: not detected.a Reaction conditions: 25.0 mg catalyst, 0.39 g p-CNB in 4.0 mL ethanol (solvent), 323 K, 1.0 MPa H2, stirring speed: 900 rpm.b 4,4′-dichloroazoxybenzene and 4,4′-dichloroazobenzene. | |||||
| Pt0.0001/TiO2 | 60 | n.d. | n.d. | n.d. | n.d. |
| Au0.005/TiO2 | 60 | 0.9 | 94.7 | n.d. | 5.3 |
| Pt0.0001–Au0.005/TiO2 | 60 | 99.2 | 99.9 | n.d. | 0.1 |
| Pt0.0002–Au0.005/TiO2 | 45 | 100 | 100 | n.d. | n.d. |
| Pt0.0003–Au0.005/TiO2 | 40 | 100 | 100 | n.d. | n.d. |
| Pt0.0004–Au0.005/TiO2 | 38 | 100 | 99.9 | 0.1 | n.d. |
| Pt0.0005–Au0.005/TiO2 | 37 | 100 | 99.5 | 0.5 | n.d. |
The effect of the Pt0.0002–Au0.005/TiO2 catalyst amount expressed as weight percent of catalyst based on p-CNB was studied in the range of 0–9% (w/w) (Fig. 1). The conversion of p-CNB varied linearly with the increase in catalyst amount up to 6.4% and then levelled off. Therefore, it appears that up to a catalyst amount of 6.4% (w/w) the reaction is mainly controlled by either the rate of mass transfer from the bulk liquid to the catalyst surface or the intrinsic kinetics of the reaction. Since, for the size of the catalyst used and the conditions employed, the diffusion of hydrogen from the bulk liquid to the solid is estimated to be unimportant, the data appears to represent the true kinetics of the process at a catalyst amount of 6.4% (w/w), temperature of 323 K and 1.0 MPa H2 for 97.5 mg mL−1 of p-CNB.
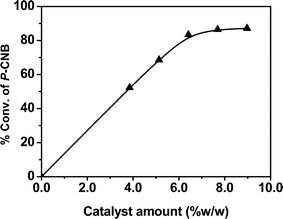 | ||
| Fig. 1 Effect of Pt0.0002–Au0.005/TiO2 catalyst amount on hydrogenation of p-CNB. 0.39 g p-CNB in 4.0 mL ethanol (solvent), 323 K, 1.0 MPa H2, 30 min, stirring speed: 900 rpm. | ||
We investigated the influence of reaction temperature on the hydrogenation of p-CNB over the Pt0.0002–Au0.005/TiO2 catalyst under 1.0 MPa H2. The results, summarized in Table 2, show that the increase in reaction temperature resulted in continuous increase in both p-CNB conversion and the selectivity to p-CAN without the formation of any decholorination product. The outstanding selectivity for p-CAN (100%) at complete conversion of p-CNB was obtained at 323–333 K. However, a hydrodechlorination process was observed with further increasing of the reaction temperature up to 343 K, and N-ethyl-p-chloroaniline, a condensation product of p-CAN with the solvent (ethanol), was also detected. Therefore, the optimum reaction temperature may be restricted to 323–333 K.
| Sel. (%) | ||||
|---|---|---|---|---|
| React. temp. (K) | p-CNB Conv. (%) | p-CAN | AN | Others |
| n.d.: not detected.a Other reaction conditions: 25.0 mg catalyst, 0.39 g p-CNB in 4.0 mL ethanol (solvent), 1.0 MPa H2, 1 h, stirring speed: 900 rpm.b 4,4′-dichloroazoxybenzene and 4,4′-dichloroazobenzene.c N-ethyl-p-chloroaniline | ||||
| 303 | 10.1 | 93.9 | n.d. | 6.1b |
| 313 | 55.2 | 97.3 | n.d. | 2.7b |
| 323 | 100 | 100 | n.d. | n.d. |
| 333 | 100 | 100 | n.d. | n.d. |
| 343 | 100 | 99.7 | 0.2 | 0.1c |
H2 pressure has a significant effect on the substrate reactivity and product selectivity (Table 3). The conversion of p-CNB and selectivity to p-CAN were seen to increase with the H2 pressure. The conversion of p-CNB and selectivity to p-CAN were 63.2% and 97.9%, respectively, at 0.5 MPa H2. The conversion increased to 100% and the selectivity reached 100% at 1.0 MPa H2. The H2 utilization rate in the reaction was ca. 89% by theoretical calculations according to the following formula: % utilization rate = m/M×4/(25×10−3×10/22.4), where m is the mass of p-CNB; M the mole mass of p-CNB. It is interesting to note that the selectivity to p-CAN could be maintained even when the reaction was conducted at 4 MPa H2. These data demonstrate that the undesired hydrodechlorination can be avoided over the Pt0.0002–Au0.005/TiO2 catalyst even when very high H2 pressure is used for the reaction.
| Sel. (%) | ||||
|---|---|---|---|---|
| H2 pressure (MPa) | p-CNB Conv. (%) | p-CAN | AN | Othersb |
| n.d.: not detected.a Other reaction conditions: 25.0 mg catalyst, 0.39 g p-CNB in 4.0 mL ethanol (solvent), 323 K, 1 h, stirring speed: 900 rpm.b 4,4′-dichloroazoxybenzene and 4,4′-dichloroazobenzene. | ||||
| 0.5 | 63.2 | 97.9 | n.d. | 2.1 |
| 1.0 | 100 | 100 | n.d. | n.d. |
| 2.0 | 100 | 100 | n.d. | n.d. |
| 4.0 | 100 | 100 | n.d. | n.d. |
The effect of the reaction time for the hydrogenation of p-CNB over Pt0.0002–Au0.005/TiO2 was studied at 323 K, 1.0 MPa H2. As shown in Table 4, both p-CNB conversion and the selectivity for p-CAN increased along with the reaction time. When the reaction was continued for 45 min, we observed a complete conversion of p-CNB and 100% selectivity for p-CAN. It is known for the catalytic reaction of interest, that a high selectivity is difficult to maintain once the substrate has been exhausted completely over most metal-supported catalysts, since in the absence of a substrate the catalytic hydrodechlorination reaction will occur at a much faster rate. To our surprise, the high selectivity could be maintained even when the reaction time was extended by several hours after p-CNB was exhausted.
| Sel. (%) | ||||
|---|---|---|---|---|
| React. time (min) | p-CNB Conv. (%) | p-CAN | AN | Others |
| n.d.: not detected.a Other reaction conditions: 25.0 mg catalyst, 0.39 g p-CNB in 4.0 mL ethanol (solvent), 323 K, 1.0 MPa H2, stirring speed: 900 rpm.b 4,4′-dichloroazoxybenzene and 4,4′-dichloroazobenzene.c N-ethyl-p-chloroaniline. | ||||
| 15 | 51.8 | 97.1 | n.d. | 2.9b |
| 30 | 83.4 | 98.7 | n.d. | 1.3b |
| 45 | 100 | 100 | n.d. | n.d. |
| 60 | 100 | 100 | n.d. | n.d. |
| 120 | 100 | 100 | n.d. | n.d. |
| 180 | 100 | 100 | n.d. | n.d. |
| 240 | 100 | 99.9 | n.d. | 0.1c |
To explore the stability of the Pt0.0002–Au0.005/TiO2 for p-CNB hydrogenation, the catalyst was separated from the reaction system after the p-CNB hydrogenation reaction had completed, it was washed with ethanol, dried under vacuum and then reused for the same reaction. After reusing it five times, the p-CNB conversion and p-CAN selectivity data obtained over this recovered catalyst were found similar to those over the fresh one (Table 5). The results suggest that the Pt0.0002–Au0.005/TiO2 catalyst is very stable and suitable for practical applications.
| Sel. (%) | ||||
|---|---|---|---|---|
| Cycle | p-CNB Conv. (%) | p-CAN | AN | Others |
| n.d.: not detected.a Reaction conditions: 25.0 mg catalyst, 0.39 g p-CNB in 4.0 mL ethanol (solvent), 323 K, 1.0 MPa H2, 45 min, stirring speed: 900 rpm.b 4,4′-dichloroazoxybenzene and 4,4′-dichloroazobenzene. | ||||
| 1 | 100 | 100 | n.d. | n.d. |
| 2 | 96.5 | 99.9 | n.d. | 0.1b |
| 3 | 95.6 | 99.9 | n.d. | 0.1b |
| 4 | 95.1 | 99.9 | n.d. | 0.1b |
| 5 | 94.8 | 99.9 | n.d. | 0.1b |
The X-ray diffraction (XRD) patterns of the TiO2, Au0.005/TiO2 and Pt0.0002–Au0.005/TiO2 catalysts are given in Fig. 2. Peak assignments for TiO2 (Fig. 2a, 2θ = 25.2, 37.8, 48.1, 53.9 and 55.1°) are consistent with the (101), (004), (200), (105) and (211) planes associated with tetragonal anatase (JCPDS-ICDD 21-1272). Apart from the diffraction peaks of the anatase TiO2, there were no peaks in the XRD patterns of the Au0.005/TiO2 (Fig. 2b) and the Pt0.0002–Au0.005/TiO2 (Fig. 2c), i.e., no new phases were detected. Moreover, neither the Au0.005/TiO2 nor the Pt0.0002–Au0.005/TiO2 presented obvious change compared to the TiO2 in the XRD spectra (Fig. 2). This result can presumably be ascribed to the combinations of low contents of doping Au and Pt below the detection limits of the XRD, small grain size, and homogeneous distribution.
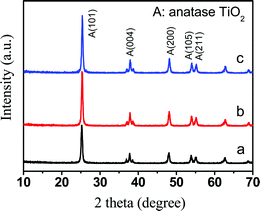 | ||
| Fig. 2 XRD patterns of TiO2 (a), Au0.005/TiO2 (b) and Pt0.0002–Au0.005/TiO2 (c). | ||
In order to obtain explicit details of Au and Pt particle size, Au0.005/TiO2 and Pt0.0002–Au0.005/TiO2 were examined with transmission electron microscopy (TEM) in the dark field mode. As can be seen from the TEM images (Fig. 3a,b), small bright spots can be discerned from the background over Au0.005/TiO2 and Pt0.0002–Au0.005/TiO2. A combined EDX analysis, using an electron beam of 0.8 nm in diameter, showed that the signal of Au could be detected only at the small bright spots (Fig. 3b1,b2). No Pt signals were observed in the EDX spectrum (Fig. 3b2). The crystal lattice spacing of a selected metal particle (Figure S1, ESI†) is rather uniform and consistent with that of Au single crystallites,24 suggesting that the Pt and Au are probably not alloyed. These are probably due to the low content of Pt in the Pt0.0002–Au0.005/TiO2 (only 0.02 wt%). Therefore, we conclude that the small bright spots in Fig. 3b are the image of the Au or Au and Pt species. The Au or Au and Pt particles in the Pt0.0002–Au0.005/TiO2 catalyst have an average diameter of 5 nm, with a size distribution from 3 to 6 nm (Fig. 3b), which is consistent with the absence of characteristic diffraction peaks of Au and Pt in the XRD pattern of Pt0.0002–Au0.005/TiO2, suggesting that the Au and Pt species on TiO2 are in a homogenous dispersion without any obvious aggregation.
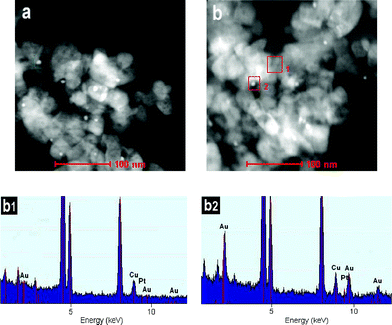 | ||
| Fig. 3 TEM images of Au0.005/TiO2 (a), Pt0.0002–Au0.005/TiO2 (b) and EDX spectra of the selected area 1 (b1) and 2 (b2). | ||
Gold particle size and distribution can have a direct impact on the hydrogenation activity where the presence of nanoscale Au particles (<10 nm) has been deemed essential for significant activity,25 with a decrease in efficiency for larger Au particles. This interpretation cannot be applied in the present work, for these Au particles in the Pt0.0002–Au0.005/TiO2 did not change obviously in size when Pt was added (Fig. 3a,b). The hydrogenation activity of the Au nanoparticles was often much lower than those of the platinum-group metals. Some observations show that the low hydrogenation activity of the Au nanoparticles is most probably due to an “intrinsic” nobleness of Au for H2activation, and that the dissociative adsorption of H2 on Au is an activated process and is restricted to the edge and corner sites or to the interface perimeter around the Au nanoparticles that are in contact with the oxide support.20 Therefore, the activity enhancement of Au nanoparticles by the deposition of Pt entities for the chemoselective hydrogenation of p-CNB could be explained by a synergic effect of Pt, Au and the TiO2 support (Fig. 4). It seems reasonable that the Pt sites were responsible for the dissociative activation of H2 molecules while Au sites, especially the perimeter interfaces around the Au particles in contact with the TiO2 support for the adsorption-activation of the substrate molecules. The activated H atoms would then migrate by spilling over toward nearby Au sites, especially perimeter interfaces between TiO2 and Au nanoparticles for a fast reaction with the adsorbed substrate. The dramatic enhancement of the Au activity by the Pt entities would suggest that the overall reaction rate was determined by the activation of H2 or by the supply of H atoms. On the other hand, the well maintained selectivity propensity of the Au nanoparticles in the hydrogenation reactions, regardless of Pt in the Pt0.0002–Au0.005/TiO2 catalyst, would indicate that such chemoselective hydrogenation selectivity was governed by the adsorption mode of the substrate molecules on the Au sites, especially perimeter interfaces between the TiO2 and Au nanoparticles.
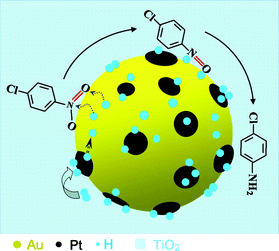 | ||
| Fig. 4 Pt promotion of the hydrogenation catalysis of Au nanoparticles. | ||
3. Conclusions
In conclusion, adding a small amount of Pt entities (0.01–0.03 wt%) onto the Au surface of a Au/TiO2 catalyst is an efficient approach to improve the catalytic activity of Au for the hydrogenation of p-chloronitrobenzene, while the C–Cl bond in the p-CNB molecule remained intact. XRD, TEM and catalytic results implied that this unusually high reaction rate and selectivity might be due to the Pt sites acting to activate the H2 molecules while the Au sites, especially perimeter interfaces around the Au particles in contact with the TiO2 support serve to activate p-CNB by chemisorption. There was a remarkable effect of the Pt loading and reaction temperature on p-CAN selectivity. Excess amounts of Pt (>0.03 wt%) and high reaction temperatures will cause the occurrence of the undesired catalytic hydrodechlorination reaction of p-CNB. As for Pt0.0002–Au0.005/TiO2, the selectivity for p-CAN could be up to 100% at complete conversion of p-CNB with reaction temperatures at or below 333 K. In addition, the catalyst was quite stable and could be used repetitively without significant deactivation. These facts allow us to view it as a prospective heterogeneous catalyst for selective production of p-CAN from p-CNB.4. Experimental
4.1 Catalyst preparation
TiO2 with a BET surface area of 42 m2 g−1 and an average grain size of 35 nm was prepared by hydrothermal homogeneous precipitation according to the following procedure: 6 g urea was added into 50 mL of 16.5% (w/v) Ti(SO4)2 aqueous solution (Beijing Chem. Co.). After stirring for 30 min at room temperature, the mixture was transferred into an 80 mL teflon-lined autoclave for hydrothermal reaction under 433 K for 20 h and cooled to room temperature naturally. The resultant precipitate was filtered and washed with distilled water until it was free from sulfate ions, then dried at 383 K overnight. Finally, the TiO2 was obtained by calcination in flowing air at 873 K for 5 h. The Pt–Au/TiO2 catalyst was prepared using HAuCl4, H2PtCl6 for the precursors of gold and platinum by deposition–precipitation with urea. Typically, 2.0 g of TiO2 was suspended in 50 mL aqueous solution of HAuCl4 (1.015 × 10−3 mol L−1), H2PtCl6 (4.1 × 10−5 mol L−1) and urea (0.2 mol L−1). The mixture was heated to 353 K and maintained at this temperature for 4 h under vigorous stirring and allowed to cool to room temperature overnight. The precipitate was isolated and washed thoroughly with distilled water by filtration until it was free from chloride ions. Finally, the sample was dried at 383 K for 12 h and calcined at 473 K for 5 h in flowing air (60 mL min−1). The amount of gold and platinum in the catalyst was 0.49 wt% and 0.018 wt%, respectively, according to ICP–AES analysis.4.2 Catalyst characterization
The gold and platinum contents in the Pt0.0002–Au0.005/TiO2 catalyst were analyzed by means of inductively coupled plasma–atomic emission spectroscopy (ICP–AES, Leeman Prodigy) after the sample was dissolved in aqua regia. X-ray diffraction (XRD) analysis was carried out on a Rigaku D/Max Ultima IV X-ray diffractometer with Cu Kα radiation (λ = 0.154056 nm). The catalyst for XRD characterization was prepared by placing some sample on a glass slide. The transmission electron microscopy (TEM) measurement was performed on a JEOL JEM-2000EX microscope operated at an accelerating voltage of 120 kV. An energy-dispersive X-ray (EDX) spectroscopic detecting unit was used to collect the EDX spectra for elemental analysis. A sample for TEM examination was made by placing a drop of the catalyst powder dispersion in ethanol on a carbon-coated copper grid, followed by drying at room temperature.4.3 Catalysis procedure
Catalytic tests were performed with magnetic stirring in a 25 mL stainless steel autoclave containing 0.39 g p-CNB in 4.0 mL ethanol (solvent). The catalyst (Pt–Au0.005/TiO2 powder, particle size 0.1 mm) used in each run of the reaction was 25.0 mg. The overall molar p-CNB/Au ratio in the reaction mixture was 3902. The reactor was flushed five times with 0.5 MPa H2 before it was pressurized to the desired H2 pressure and placed into an oil bath maintained at the reaction temperature. The reaction was stopped at a selected time by cooling the reactor in an ice–water bath. Analysis of the reaction products was done on an HP6890A GC instrument with a HP-5 capillary column with a FID detector, using benzene as an internal standard.Acknowledgements
This work was financially supported by the Chongqing Education Commission (KJ110621), the Chongqing Normal University (10XLR021).Notes and references
- A. S. Travis, in The Chemistry of Anilines, ed. Z. Rappoport, John Wiley & Sons, Ltd, Chichester, England, 2007, pp. 715–782 Search PubMed.
- H. U. Blaser, U. Siegrist, H. Steiner and M. Studer, in Fine Chemicals through Heterogeneous Catalysis, Wiley-VCH, Weinheim, 2001, pp. 389–406 Search PubMed.
- K. R. Westerterp, K. B. van Gelder, H. J. Janssen and M. H. Oyevaar, Chem. Eng. Sci., 1988, 43, 2229–2236 CrossRef CAS; B. Coq, A. Tijani and F. Figueras, J. Mol. Catal., 1991, 68, 331–345 CrossRef.
- Y. Z. Chen and Y. C. Chen, Appl. Catal., A, 1994, 115, 45–47 CrossRef CAS; V. L. Khilnani and S. B. Chandalia, Org. Process Res. Dev., 2001, 5, 257–262 CrossRef; H. Q. Liu, M. H. Liang, C. Xiao, N. Zheng, X. H. Feng, Y. Liu, J. L. Xie and Y. Wang, J. Mol. Catal. A: Chem., 2009, 308, 79–86 CrossRef.
- X. X. Han, R. X. Zhou, X. M. Zheng and H. Jiang, J. Mol. Catal. A: Chem., 2003, 193, 103–108 CrossRef CAS.
- X. X. Han, R. X. Zhou, G. H. Lai, B. H. Yue and X. M. Zheng, J. Mol. Catal. A: Chem., 2004, 209, 83–87 CrossRef CAS.
- W. X. Tu, H. F. Liu and Y. Tang, J. Mol. Catal. A: Chem., 2000, 159, 115–120 CrossRef CAS; M. H. Liu, J. Zhang, J. Q. Liu and W. W. Yu, J. Catal., 2011, 278, 1–7 CrossRef.
- Y. C. Liu, C. Y. Huang and Y. W. Chen, Ind. Eng. Chem. Res., 2006, 45, 62–69 CrossRef CAS; H. Li, Q. F. Zhao and H. X. Li, J. Mol. Catal. A: Chem., 2008, 285, 29–35 CrossRef.
- B. J. Zuo, Y. Wang, Q. L. Wang, J. L. Zhang, N. Z. Wu, L. D. Peng, L. L. Gui, X. D. Wang, R. M. Wang and D. P. Yu, J. Catal., 2004, 222, 493–498 CrossRef CAS.
- G. C. Bond, C. Louis and D. T. Thompson, Catalysis by Gold, Imperical College Press, London, 2006, pp.161-356 CrossRef CAS; M. Haruta, Gold Bull., 2004, 37, 27–36 CrossRef CAS.
- M. Haruta, N. Yamada, T. Kobayashi and S. Iijima, J. Catal., 1989, 115, 301–309 CrossRef CAS; C. Rossignol, S. Arrii, F. Morfin, L. Piccolo, V. Caps and J. L. Rousset, J. Catal., 2005, 230, 476–483 CrossRef.
- C. H. Christensen, B. Jorgensen, J. Rass-Hansen, K. Egeblad, R. Madsen, S. K. Klitgaard, S. M. Hansen, M. R. Hansen, H. C. Andersen and A. Riisager, Angew. Chem., Int. Ed., 2006, 45, 4648–4651 CrossRef CAS; R. L. Oliveira, P. K. Kiyohara and L. M. Rossi, Green Chem., 2010, 12, 144–149 RSC.
- M. D. Hughes, Y. J. Xu, P. Jenkins, P. McMorn, P. Landon, D. I. Enache, A. F. Carley, G. A. Attard, G. J. Hutchings, F. King, E. H. Stitt, P. Johnston, K. Griffin and C. J. Kiely, Nature, 2005, 437, 1132–1135 CrossRef CAS; S. Bawaked, N. F. Dummer, N. Dimitratos, D. Bethell, Q. He, C. J. Kiely and G. J. Hutchings, Green Chem., 2009, 11, 1037–1044 RSC.
- B. Nkosi, N. J. Coville, G. J. Hutchings, M. D. Adams, J. Friedl and F. E. Wagner, J. Catal., 1991, 128, 366–377 CrossRef CAS; D. T. Thompson, Appl. Catal., A, 2003, 243, 201–205 CrossRef.
- M. Okumura, T. Akita and M. Haruta, Catal. Today, 2002, 74, 265 CrossRef CAS; X. Zhang, H. Shi and B. Q. Xu, Angew. Chem., Int. Ed., 2005, 44, 7132–7135 CrossRef; X. F. Yang, A. Q. Wang, Y. L. Wang, T. Zhang and J. Li, J. Phys. Chem. C, 2010, 114, 3131–3139 Search PubMed.
- C. Milone, R. Ingoglia, A. Pistone, G. Neri, F. Frusteri and S. Galvagno, J. Catal., 2004, 222, 348–356 CrossRef CAS; Y. C. Hong, K. Q. Sun, G. R. Zhang, R. Y. Zhong and B. Q. Xu, Chem. Commun., 2011, 47, 1300–1302 RSC.
- A. Corma and P. Serna, Science, 2006, 313, 332–333 CrossRef CAS; Y. Y. Chen, J. S. Qiu, X. K. Wang and J. H. Xiu, J. Catal., 2006, 242, 227–230 CrossRef; F. Cárdenas-Lizana, S. Gómez-Quero and M. A. Keane, ChemSusChem, 2008, 1, 215–221 CrossRef; P. Serna, M. Boronat and A. Corma, Top. Catal., 2011, 54, 439–446 CrossRef.
- D. P. He, H. Shi, Y. Wu and B. Q. Xu, Green Chem., 2007, 9, 849–851 RSC.
- G. Vitulli, A. Verrazzani, E. Pitzalis, P. Salvadori, G. Capannelli and G. Martra, Catal. Lett., 1997, 44, 205–210 CrossRef CAS; H. W. Ma, K. P. Sun, Y. W. Li and X. L. Xu, Catal. Commun., 2009, 10, 1363–1366 CrossRef.
- E. Bus, J. T. Miller and J. A. van Bokhoven, J. Phys. Chem. B, 2005, 109, 14581–14587 CrossRef CAS; T. Fujitani, L. Nakamura, T. Akita, M. Okumura and M. Haruta, Angew. Chem., Int. Ed., 2009, 48, 9515–9518 CrossRef.
- V. Kratky, M. Kralik, M. Mecarova, M. Stolcova, L. Zalibera and M. Hronec, Appl. Catal., A, 2002, 235, 225–231 CrossRef CAS; J. Xiong, J. X. Chen and J. Y. Zhang, Catal. Commun., 2007, 8, 345–350 CrossRef; J. X. Chen, N. Yao, R. J. Wang and J. Y. Zhang, Chem. Eng. J., 2009, 148, 164–172 CrossRef.
- Y. C. Liu and Y. W. Chen, Ind. Eng. Chem. Res., 2006, 45, 2973–2980 CrossRef CAS; C. L. Fernando, G. Q. Santiago and A. K. Mark, Appl. Catal., A, 2008, 334, 199–206 CrossRef.
- S. Galvagno, A. Donato, G. Neri and R. Poetrpaolo, J. Mol. Catal., 1987, 42, 379–387 CrossRef CAS.
- P. Claus, H. Hofmeister and C. Mohr, Gold Bull., 2004, 37, 181–186 CrossRef CAS.
- M. Haruta, Catal. Today, 1997, 36, 153–166 CrossRef CAS; C. Mohr, H. Hofmeister and P. Claus, J. Catal., 2003, 213, 86–94 CrossRef; R. Zanella, C. Louis, S. Giorgio and R. Touroude, J. Catal., 2004, 223, 328–339 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: See DOI: 10.1039/c1gc16001j |
| This journal is © The Royal Society of Chemistry 2012 |