Near-critical water, a cleaner solvent for the synthesis of a metal–organic framework†
Ilich A.
Ibarra
a,
Peter A.
Bayliss
a,
Eduardo
Pérez
a,
Sihai
Yang
a,
Alexander J.
Blake
a,
Harriott
Nowell
b,
David R.
Allan
b,
Martyn
Poliakoff
*a and
Martin
Schröder
*a
aSchool of Chemistry, The University of Nottingham, Nottingham, UK NG7 2RD. E-mail: Martyn.Poliakoff@nottingham.ac.uk; M.Schroder@nottingham.ac.uk
bDiamond Light Source, Harwell Science and Innovation Campus, Didcot, Oxfordshire, UK OX11 0DE
First published on 14th November 2011
Abstract
The microporous metal–organic framework {[Zn2(L)]·(H2O)3}∞ (H4L = 1,2,4,5-tetrakis(4-carboxyphenyl)benzene) has been synthesised using near-critical water (300 °C) as a cleaner alternative to toxic organic solvents. A single crystal X-ray structure determination confirms that the complex incorporates tetrahedral Zn(II) centres bridged through the carboxylate anions to form a binuclear building block, which extends into a one dimensional chain along the c axis. Four L4− ligands bind to each Zn(II) centre and cross-link the one dimensional chains along both a and b axes to afford a three dimensional network structure incorporating pores of ca. 4.3 Å in diameter. The complex shows high thermal stability up to 425 °C by gravimetric thermal analysis, and on desolvation, displays a high adsorption enthalpy of 11.0 kJ mol−1 for H2 uptake at zero coverage, consistent with the narrow pore diameter for the framework.
Introduction
Metal–organic frameworks (MOFs) are novel materials based upon combinations of metal centres and polychelate bridging ligands. MOFs are receiving increasing attention due to their interesting properties,1 and their potential applications in gas storage2 and separation,3 catalysis,4 optics5 and magnetism.6 There is widespread interest in the use of MOFs for hydrogen (H2) storage,7 but a major challenge remains to synthesise porous storage materials that operate at ambient temperatures. In this context, an important parameter to consider is the isosteric heat of adsorption,8 which defines the binding energy of physisorption of H2 in porous materials at different loadings. Typical values for the heat of adsorption of H2 binding in MOFs at zero loading are between 5–7 kJ mol−1,9–11 which means that these materials need to operate at low temperatures (typically 77 K) to afford respectable storage capacities. A higher isosteric heat of adsorption of some 20 kJ mol−1 is required to achieve efficient H2storage at ambient temperatures.12 A number of strategies have been adopted to increase the H2 binding energy in these materials including focus on the formation of narrow pores10,11,13 and access to exposed metal sites.9,14In this paper we focus on finding a potentially cleaner route to making a MOF from a metal salt plus a ligand rather than addressing the synthesis of the ligand itself. This is because no one has yet identified the specific metal/ligand combination to give a MOF with the properties necessary for viable H2storage. However, the general procedures for synthesising MOFs are sufficiently well established to justify trying to make these procedures more sustainable. These currently involve quantities of solvent that are large compared to the yield of product and any sustainable approach should involve a solvent that has low hazard and is simple to recycle.
MOF materials are often prepared using solvothermal routes15 where the component metal salt and ligand are reacted in organic solvents or solvent mixes typically composing alkyl formamides,16alcohols17 or pyrrolidones.18 Heating of reaction solutions in sealed vessels, such as Teflon-lined stainless steel bombs or glass pressure tubes, often affords products that incorporate solvent molecules bound to the metal centres as well as being present within the pores. These organic solvents are often toxic, carcinogenic and/or environmentally hazardous. For example, dimethylformamide (DMF), a solvent very commonly used in MOF synthesis, is a pollutant, a mutagen and is toxic.19 In addition, DMF decomposes when heated to high temperatures for long periods and therefore cannot be reused.20 Thus, concern over the use of such organic solvents is increasing due to their negative environmental impact,21 and replacing the most common solvents in MOFs synthesis with more benign ones offers an opportunity to apply the principles of green chemistry22 to this technology.
Our starting point is the well-established hydrothermal synthesis23 of MOFs, which involves mixing components with a small amount of water and heating in a sealed vessel typically up to 200 °C. In this paper we exploit the properties of high-temperature water (HTW), which is being studied as a medium for carrying out processes such as organic reactions,24 waste destruction25 and nanoparticle formation.26HTW is an interesting solvent because the properties of water change dramatically as it approaches the critical point (374 °C, 220 bar).27 For example, the dielectric constant decreases to values more typical of non-polar solvents and therefore organic compounds, such as the extended ligands used for the synthesis of MOFs, become soluble. Furthermore, the ionic product of the reaction solution increases by up to three orders of magnitude reaching a maximum at around 280 °C, thereby creating a solvent that has simultaneously raised concentrations of H+ and OH− ions.
Water can potentially be re-used in the reaction without purification and, if necessary, processes such as ion exchange can be used to remove unreacted carboxylic acid and metal ions. Until now, the technical difficulties presented by HTW and supercritical H2O, particularly high pressures and accelerated rates of corrosion, appear to have discouraged chemists from exploring the potential of these solvents for the synthesis of MOFs.
We report herein for the first time the feasibility of using near-critical water (300 °C) as a solvent for the synthesis of MOFs in high yield, and present the synthesis of a new Zn(II)-carboxylate material using only H2O as the reaction medium. One advantage of using Zn(II) is that it is less toxic than many transition metals. The desolvated product shows permanent porosity and high isosteric heat of adsorption for H2storage. The synthesis of the co-ordination polymer was carried out in a high-pressure stainless steel batch reactor28 using water as solvent with the temperature set to 300 °C, which generated a pressure of 80 bar.
Results and discussion
Reaction of Zn(NO3)2 with 1,2,4,5-tetrakis(4-carboxyphenyl)benzene (H4L) (Scheme 1), prepared via Suzuki coupling of 1,2,4,5-tetrabromobenzene and 4-ethoxycarbonylphenylboronic acid, for 48 h in HTW at 300 °C produced {[Zn2(L)]·(H2O)3}∞ in 78% yield based on H4L. The single crystal X-ray structure determination confirms that the complex crystallises in the monoclinic space groupC2/c with each tetrahedral Zn(II) centre co-ordinated to four O-centres from four different carboxylate groups, Zn–O = 1.910(10) and 1.921(10) Å. The Zn(II) centres are bridged through the carboxylate anions to form a binuclear building block (Fig. 1), which extends into a one dimensional chain along the c axis with Zn⋯Zn separations of 3.423(5) and 3.449(5) Å (Fig. 2a). Four L4− ligands bind to each Zn(II) centre and cross-link the one dimensional chains along both a and b axes (Fig. 2b) to afford a three dimensional network structure incorporating pores of ca. 4.3 Å in diameter, with a crystallographic pore volume calculated by PLATON29 of 0.25 cm3 g−1 using a default probe size of 1.2 Å (Fig. 3). Significantly, the complex does not incorporate H2O molecules bound directly to Zn(II), but H2O molecules do reside within the pores. Thus, even though high temperature water has been used as the reaction medium, the product does not contain co-ordinated water as a ligand. The use of higher temperatures in solvothermal synthesis to reduce water content in a series of cobalt succinate materials has been reported previously,30 while a similar topology to that in {[Zn2(L)]·(H2O)3}∞ has been previously observed for [Zn2(1,4-dihydroxy-2,5-benzenediphosphonate)](DMF)2.31 The analogous solvothermal reaction of Zn(II) with H4L carried out in DMF affords the corresponding DMF-solvated species [Zn2(L)(DMF)2]∞, which shows a different structure to that of {[Zn2(L)]·(H2O)3}∞ and incorporates a binuclear paddlewheel node [Zn2(O2CR)4].31 Interestingly, Zn(NO3)2 reacted with H4L in HTW under the same conditions as above but using 150 °C for 48 h does not afford the same product as obtained when running the reaction at 300 °C; this unidentified product exhibits poor crystallinity and its powder X-ray diffraction (PXRD) pattern does not match the pattern shown by {[Zn2(L)]·(H2O)3} (see ESI†).![A view of the binuclear building block in {[Zn2(L)]·(H2O)3}∞ formed by L4− co-ordinated to two Zn(ii) centres; (zinc: green; oxygen: red; carbon: black; hydrogen: small grey).](/image/article/2012/GC/c1gc15726d/c1gc15726d-f1.gif) | ||
| Fig. 1 A view of the binuclear building block in {[Zn2(L)]·(H2O)3}∞ formed by L4− co-ordinated to two Zn(II) centres; (zinc: green; oxygen: red; carbon: black; hydrogen: small grey). | ||
![(left a) A view of the one dimensional RCO2-bridged Zn(ii) chain along the c axis, (atom colours are as in Fig. 1), and (right b) view of the orthogonal phenyl groups of the four [L]−4 ligands co-ordinated to each Zn(ii) centre showing the generation of the three dimensional network structure.](/image/article/2012/GC/c1gc15726d/c1gc15726d-f2.gif) | ||
| Fig. 2 (left a) A view of the one dimensional RCO2-bridged Zn(II) chain along the c axis, (atom colours are as in Fig. 1), and (right b) view of the orthogonal phenyl groups of the four [L]−4 ligands co-ordinated to each Zn(II) centre showing the generation of the three dimensional network structure. | ||
![A space-filling view of the structure of {[Zn2(L)]·(H2O)3}∞ along the b axis showing the 4.3 Å channels; (atom colours are as in Fig. 1).](/image/article/2012/GC/c1gc15726d/c1gc15726d-f3.gif) | ||
| Fig. 3 A space-filling view of the structure of {[Zn2(L)]·(H2O)3}∞ along the b axis showing the 4.3 Å channels; (atom colours are as in Fig. 1). | ||
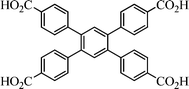 | ||
| Scheme 1 A view of H4L. | ||
Thermogravimetric analysis of {[Zn2(L)]·(H2O)3}∞ under N2 shows a weight loss of ca. 7.0% up to 425 °C attributed to the loss of solvent (H2O) (theoretical amount 7.3%) with the material decomposing at temperatures above 425 °C (Fig. 4). Good correlation between the experimental PXRD data with that calculated from the single crystal data was obtained (Fig. 5) while in situ variable-temperature PXRD under vacuum confirmed retention of crystallinity of the [Zn2(L)] framework upon desolvaton up to 300 °C (Fig. 6). The unco-ordinated H2O molecules in the pores of the framework {[Zn2(L)]·(H2O)3}∞ can be exchanged for other organic solvents, such as acetone, to promote the formation of the solvent-free framework [Zn2(L)] on heating. Thus, {[Zn2(L)]·(H2O)3}∞ was treated with acetone for 96 h at room temperature and subsequently degassed at 170 °C and 10−10 bar for 12 h to isolate the fully desolvated material [Zn2(L)]. N2 adsorption in [Zn2(L)] at 77 K shows a Type I isotherm (Fig. 7a) characteristic of a microporous material, with a BET surface area of 285 m2 g−1 and a total micropore volume of 0.20 cm3 g−1, consistent with 0.25 cm3 g−1 calculated from the single crystal structure using PLATON.29 The observed hysteresis loop with a step in the profile for N2 adsorption–desorption is evidence of a kinetic trap33 consistent with the restricted dimensions of the pores. From the N2 isotherm the pore size distribution of desolvated [Zn2(L)] was estimated to be 4.5 Å using the DR equation (see ESI†) and this value is in good agreement with that estimated from the single crystal structure (4.3 Å).
![Thermogravimetric analysis of {[Zn2(L)]·(H2O)3}∞.](/image/article/2012/GC/c1gc15726d/c1gc15726d-f4.gif) | ||
| Fig. 4 Thermogravimetric analysis of {[Zn2(L)]·(H2O)3}∞. | ||
![Experimental PXRD pattern and simulated pattern derived from the single crystal structure of {[Zn2(L)]·(H2O)3}∞.](/image/article/2012/GC/c1gc15726d/c1gc15726d-f5.gif) | ||
| Fig. 5 Experimental PXRD pattern and simulated pattern derived from the single crystal structure of {[Zn2(L)]·(H2O)3}∞. | ||
![Variable temperature in situPXRD patterns for {[Zn2(L)]·(H2O)3}∞.](/image/article/2012/GC/c1gc15726d/c1gc15726d-f6.gif) | ||
| Fig. 6 Variable temperature in situPXRD patterns for {[Zn2(L)]·(H2O)3}∞. | ||
![(left a) N2 isotherm for desolvated [Zn2(L)] 0–1 bar and 77 K; (centre b) H2 isotherm up to 20 bar for desolvated [Zn2(L)] at 77 K and (right c) variation of the adsorption enthalpy at low loading for [Zn2(L)].](/image/article/2012/GC/c1gc15726d/c1gc15726d-f7.gif) | ||
| Fig. 7 (left a) N2 isotherm for desolvated [Zn2(L)] 0–1 bar and 77 K; (centre b) H2 isotherm up to 20 bar for desolvated [Zn2(L)] at 77 K and (right c) variation of the adsorption enthalpy at low loading for [Zn2(L)]. | ||
H2 adsorption experiments were carried out on desolvated [Zn2(L)] from 0–20 bar at 77 K. All data were rigorously corrected for the buoyancy of systems, samples and absorbates. The H2 sorption isotherm shows good reversibility, characteristic of microporous materials. The H2 isotherm shows slight hysteresis reflecting the very narrow pores in [Zn2(L)]; the hysteresis loop is more clearly defined for N2, which has a larger kinetic diameter (3.64 Å) than H2 (2.89 Å). The total H2 uptake for [Zn2(L)] is recorded as 1.64 wt% at 20 bar and 77 K (Fig. 7b).
The heat of adsorption of H2 in [Zn2(L)] was determined by recording H2 isotherms at 77 and 87 K at pressures of 0–20 bar. These data were analysed using a virial-type equation,34 and the calculated isosteric heat of adsorption at zero surface coverage (Fig. 7c) was found to be 11.0 kJ mol−1. The pores within this framework are narrow (4.5 Å diameter estimated from the N2 adsorption and 4.3 Å diameter estimated from the single crystal X-ray structure) and such narrow pores enhance the H2 binding energy in porous materials at low loadings.13
Conclusions
In summary, the new porous co-ordination polymer {[Zn2(L)]·(H2O)3}∞ has been successfully synthesised in near-critical water and has been fully characterised. Significantly, the complex is isolated without water bound to the Zn(II) centres contrasting with solvothermal synthesis in DMF that affords [Zn2(L)(dmf)2].32 The complex shows high thermal stability up to 300 °C. Interestingly, desolvated [Zn2(L)] is microporous with narrow pores, and thus exhibits a high heat of adsorption for H2 (11.0 kJ mol−1) at zero surface coverage reflecting the effective overlapping potential field of the pore walls.The synthesis of MOFs in near critical water represents a significant broadening of the synthetic strategies available for the construction of co-ordination polymers in an environmentally friendly way without the use of polar toxic solvents. In any industrial application, solvents cannot be reused indefinitely; at some point they require purification. Unlike organic solvents the purification of water need not necessarily involve distillation with its associated energy cost. Water does not decompose or react with the HNO3 liberated in this reaction. The acid can also be neutralised with Zn(OH)2 to generate more labile Zn(NO3)2, and excess Zn(II) in water can be removed by filtration through clays or other methods.
Experimental
All reagents and solvents were used as received from commercial suppliers without further purification. Elemental microanalyses for C, H, and N were carried out on a CE-440 elemental analyzer at the School of Chemistry, University of Nottingham. PXRD data were collected under ambient conditions and in situ under vacuum on a Bruker AXS D8 Advance diffractometer operated at 160 W (40 kV, 40 mA) for Cu-Kα1 (λ = 1.5406 Å), with temperature rate of 2 °C min−1 and step size of 0.04° in 2θ. An MRI TCP-20 with a PtRh strip heater was used for the in situ variable temperature powder X-ray diffraction experiments. Thermal gravimetric analysis (TGA) was performed under N2 atmosphere at a scan rate of 2 °C min−1 using a TA Universal Analysis 2000 system. N2 adsorption was carried out volumetrically using a Micromeritics ASAP 2020 sorptometer. The surface area was calculated using the BET method based on adsorption data in the partial pressure (P/P0) range 0.06–0.22. The pore size distribution (PSD) was determined via the Dubinin–Astakhov method applied to the nitrogen adsorption isotherm data.Synthesis of H4L
1,2,4,5-Tetrakis(4-carboxyphenyl)benzene (H4L) was synthesised via Suzuki coupling of 1,2,4,5-tetrabromobenzene and 4-ethoxycarbonylphenylboronic acid (Scheme 2). 1,2,4,5-Tetrabromobenzene (1.01 g, 2.54 mmol), 4-ethoxycarbonylphenylboronic acid (2.42 g, 11.5 mol) and K3PO4 (2.70 g, 12.70 mmol) were added to 1,4-dioxane (100 mL) under Ar. [Pd(PPh3)4] 0.0881 g, 0.08 mmol) was added to the mixture and heated to 85 °C for 4 days under Ar. The solution was evaporated to dryness and extracted with CHCl3. The CHCl3 was removed by evaporation and the final product H4L was obtained by hydrolysis of the crude product under reflux overnight with 2 M aqueous NaOH (200 mL) followed by acidification with HCl (conc.) (1.23 g, 86% yield).1H-NMR (DMSO, 300 MHz, 298 K), δH 8.08 (8H, d, Ar–H), δH 7.83 (8H, d, Ar–H), δH 7.80 (2H, s, Ar–H) ppm.13C-NMR (DMSO, 100 MHz, 298 K), δC 167.27, δC 146.45, δC 135.87, δC 128.46, δC 127.31, δC 126.94 δC 126.48 ppm. Elemental analysis (CHN) Anal. calcd (found) for H4L, C34H22O8: C 69.89% (69.92%), H 4.29% (4.38%) N 0.00% (0.00%). MS (ESI−, m/z): 557.12 (M–H)+.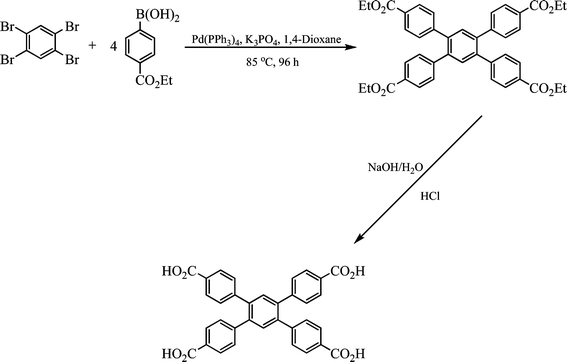 | ||
| Scheme 2 The synthesis of H4L. | ||
Synthesis of {[Zn2(L)]·(H2O)3}∞
The synthesis of {[Zn2(L)]·(H2O)3}∞ in water was carried out in a high-pressure batch reactor made of stainless steel with an internal volume of ca. 10 mL.28 The maximum operating pressure was 60 MPa at 400 °C with the reactor located in an aluminium heating block surrounded by an electric ring furnace. The temperature of the heating block was controlled by a CAL CONTROLS 3300 temperature controller, and the temperature inside the reactor was measured by a K-type thermocouple. The pressure was not controlled and was dependent on both the temperature and the contents of the cell. Internal pressure was measured using a pressure transducer. Zn(NO3)2·6H2O (110 mg, 0.36 mmol), H4L (67 mg, 0.12 mmol) and H2O (5 mL) were transferred into the reactor, and the reaction mixture was heated up to 300 °C at a rate of 45 °C h−1 and kept at that temperature for 48 h. The pressure at 300 °C within the cell was measured to be 80 ± 2 bar for the duration of the experiment. After 48 h the cell was cooled to room temperature at 25 °C h−1. The resulting brown plate shaped single crystals were recovered by filtration, washed with H2O (10 mL) and acetone (10 mL) to yield {[Zn2(C34H18O8)]·(H2O)3}∞ (67 mg, yield 78%). Elemental analysis (CHN) Anal. calcd (found): C 55.54% (55.87%), H 3.29% (3.19%), N 0.00% (0.00%).Single crystal X-ray determination
Diffraction data for {[Zn2(L)]·(H2O)3}∞ were collected on Beamline I19 at Diamond Light Source.‡ The details for data collection are included in CIF files in the Supplementary Information.† The structure was solved by direct methods and developed by difference Fourier techniques using the SHELXTL software package.35Hydrogen atoms on the ligand were placed geometrically and refined using a riding model. The unit cell volumes include a large region of disordered solvent that could not be modelled as discrete atomic sites. We employed PLATON/SQUEEZE36 to calculate the contribution to the diffraction from the solvent region and thereby produced a set of solvent-free diffraction intensities. The final formula was calculated from the SQUEEZE36 results combined with elemental analysis data.Nitrogen and hydrogen adsorption isotherms
N2 and H2 isotherms were recorded on an IGA system (Hidden Isochema, Warrington, UK) at the University of Nottingham under ultra high vacuum in a clean system with a diaphragm and turbo pumping system. Ultra-pure grade (99.9995%) H2 was purchased from BOC and purified further using calcium aluminosilicate and activated carbon adsorbents to remove trace amounts of water and other impurities before introduction into the IGA system. The density of H2 at 77 K in the buoyancy correction was calculated by the Redlich–Kwong–Soave equation of state of H2 incorporated in IGASWIN software of the IGA system.Acknowledgements
We thank the EPSRC and the University of Nottingham for support and funding, and Diamond Light Source for access to Beamline I19. MS gratefully acknowledges receipt of an ERC Advanced Grant. IAI thanks CONACYT Mexico for scholarship funding. SY thanks the University of Nottingham/EPSRC for a PhD Plus Fellowship.Notes and References
- J. L. C. Rowsell and O. M. Yaghi, Microporous Mesoporous Mater., 2004, 73, 3–14 CrossRef CAS; C. Janiak, Dalton Trans., 2003, 2781–2804 RSC; S. Kitagawa, R. Kitaura and S. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334–2375 CrossRef CAS; A. U. Czaja, N. Trukhanb and U. Müller, Chem. Soc. Rev., 2009, 38, 1284–1293 RSC.
- R. E. Morris and P. S. Wheatley, Angew. Chem., Int. Ed., 2008, 47, 4966–4981 CrossRef; M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. O'Keeffe and O. M. Yaghi, Science, 2002, 295, 469–472 CrossRef; P. L. Llewellyn, S. Bourrelly, C. Serre, A. Vimont, M. Daturi, L. Hamon, G. De Weireld, J.-S. Chang, D.-Y. Hong, Y. K. Hwang, S. H. Jhung and G. Férey, Langmuir, 2008, 24, 7245–7250 CrossRef.
- J.-R. Li, R. J. Kuppler and H.-C. Zhou, Chem. Soc. Rev., 2009, 38, 1477–1504 RSC.
- J. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450–1459 RSC; L. Ma and W. Lin, Top. Curr. Chem., 2010, 293, 175–205 CAS.
- L. Zhang, Y. Y. Qin, Z. J. Li, Q. P. Lin, J. K. Cheng, J. Zhang and Y. G. Yao, Inorg. Chem., 2008, 47, 8286–8293 CrossRef CAS.
- K. Mohamedally, Chem. Soc. Rev., 2009, 38, 1353–1379 RSC; N. Roques, V. Mugnaini and J. Veciana, Top. Curr. Chem., 2010, 293, 207–258 CAS.
- X. Lin, N. R. Champness and M. Schröder, Top. Curr. Chem., 2010, 293, 35–76 CAS; L. J. Murray, M. Dincă and J. R. Long, Chem. Soc. Rev., 2009, 38, 1294–1314 RSC; X. Lin, J. H. Jia, P. Hubberstey, M. Schröder and N. R. Champness, CrystEngComm, 2007, 9, 438–448 RSC.
- M. Dincă and J. R. Long, Angew. Chem., Int. Ed., 2008, 47, 6766–6779 CrossRef CAS; X. Lin, I. Telepeni, A. J. Blake, A. Dailly, C. M. Brown, J. M. Simmons, M. Zoppi, G. S. Walker, K. M. Thomas, T. J. Mays, P. Hubberstey, N. R. Champness and M. Schröder, J. Am. Chem. Soc., 2009, 131, 2159–2171 CrossRef CAS.
- M. Dincă and J. R. Long, J. Am. Chem. Soc., 2007, 129, 11172–11176 CrossRef CAS; Y. L. Liu, J. F. Eubank, A. J. Cairns, J. Eckert, V. C. Kravtsov, R. Luebke and M. Eddaoudi, Angew. Chem., Int. Ed., 2007, 46, 3278–3283 CrossRef CAS; M. Dincă and J. R. Long, J. Am. Chem. Soc., 2005, 127, 9376–9377 CrossRef CAS.
- W. Yang, X. Lin, J. Jia, A. J. Blake, C. Wilson, P. Hubberstey, N. R. Champness and M. Schröder, Chem. Commun., 2008, 359–361 RSC.
- Y. Liu, H. Kabbour, C. M. Brown, D. A. Neumann and C. C. Ahn, Langmuir, 2008, 24, 4772–4777 CrossRef CAS.
- S. K. Bhatia and A. L. Myers, Langmuir, 2006, 22, 1688–1700 CrossRef CAS.
- H. Chun, D. N. Dybtsev, H. Kim and K. Kim, Chem.–Eur. J., 2005, 11, 3521–3529 CrossRef CAS.
- M. Dincă, A. Dailly, Y. Liu, C. M. Brown, D. A. Neumann and J. R. Long, J. Am. Chem. Soc., 2006, 128, 16876–16883 CrossRef CAS; X. Lin, J. H. Jia, X. B. Zhao, K. M. Thomas, A. J. Blake, G. S. Walker, N. R. Champness, P. Hubberstey and M. Schröder, Angew. Chem., Int. Ed., 2006, 45, 7358–7364 CrossRef; J. Jia, X. Lin, C. Wilson, A. J. Blake, N. R. Champness, P. Hubberstey, G. Walker, E. J. Cussen and M. Schröder, Chem. Commun., 2007, 840–842 RSC; Y. Yan, X. Lin, S. Yang, A. J. Blake, A. Dailly, N. R. Champness, P. Hubberstey and M. Schröder, Chem. Commun., 2009, 1025–1027 RSC; I. A. Ibarra, X. Lin, S. Yang, A. J. Blake, G. S. Walker, S. A. Barnett, D. R. Allan, N. R. Champness, P. Hubberstey and M. Schröder, Chem.–Eur. J., 2010, 16, 13671–13679 CrossRef CAS; Y. Yan, I. Telepeni, S. Yang, X. Lin, W. Kockelmann, A. Dailly, A. J. Blake, W. Lewis, G. S. Walker, D. R. Allan, S. A. Barnett, N. R. Champness and M. Schröder, J. Am. Chem. Soc., 2010, 132, 4092–4094 CrossRef CAS; C. Tan, S. Yang, X. Lin, A. J. Blake, W. Lewis, N. R. Champness and M. Schröder, Chem. Commun., 2011, 47, 4487–4489 RSC.
- R. I. Walton, Chem. Soc. Rev., 2002, 31, 230–238 RSC; G. Demazeau, High Pressure Res., 2007, 27, 173–177 Search PubMed.
- J. Kim, B. Chen, T. M. Reineke, H. Li, M. Eddaoudi, D. B. Moler, M. O'Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2001, 34, 8239–8247 Search PubMed; O. K. Farha, C. D. Malliakas, M. G. Kanatzidis and J. T. Hupp, J. Am. Chem. Soc., 2010, 132, 950–952 CrossRef CAS; D. Sun, S. Ma, Y. Ke, D. J. Collins and H-C. Zhou, J. Am. Chem. Soc., 2006, 128, 3896–3897 CrossRef CAS; L. Ma and W. Lin, J. Am. Chem. Soc., 2008, 130, 13834–13835 CrossRef CAS; J-R. Li, A. A. Yakovenko, W. Lu, D. J. Timmons, W. Zhuang, D. Yuan and H-C. Zhou, J. Am. Chem. Soc., 2010, 132, 17599–17610 CrossRef CAS.
- M. Hartmann, S. Kunz, D. Himsl and O. Tangermann, Langmuir, 2008, 24, 8634–8642 CrossRef CAS; H. Guo, G. Zhu, I. J. Hewitt and S. Qiu, J. Am. Chem. Soc., 2009, 131, 1646–1647 CrossRef CAS.
- W. J. Son, J. Kim, J. Kim and S. Ahn, Chem. Commun., 2008, 6336–6338 RSC.
- R. P. Pohanish, Sittig's Handbook of Toxic and Hazardous Chemicals and Carcinogens, William Andrew Publishing, 5th edn, 2008 Search PubMed.
- J. Muzart, Tetrahedron, 2009, 65, 8313–8323 CrossRef CAS.
- Y. Tateyama, Y. Ohki, Y. Suzuki and A. Ouchi, Bull. Chem. Soc. Jpn., 1988, 61, 2214–2216 Search PubMed.
- P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, Oxford University Press, 1998 Search PubMed.
- O. M. Yaghi and H. Li, J. Am. Chem. Soc., 1995, 117, 10401–10402 CrossRef CAS; D. M. Young, U. Geiser, A. J. Schultz and H. H. Wang, J. Am. Chem. Soc., 1998, 120, 1331–1332 CrossRef CAS; R-G. Xiong, X. Xue, H. Zhao, X-Z. You, B. F. Abrahams and Z-L. Xue, Angew. Chem., Int. Ed., 2002, 41, 3800–3803 CrossRef CAS; X-Z. Zhang, R-Q. Fang and H-S. Wu, J. Am. Chem. Soc., 2005, 127, 7670–7671 CrossRef CAS; X. Lin, A. J. Blake, C. Wilson, X. Z. Sun, N. R. Champness, M. W. George, P. Hubberstey, R. Mokaya and M. Schröder, J. Am. Chem. Soc., 2006, 128, 10745–10753 CrossRef CAS.
- P. E. Savage, Chem. Rev., 1999, 99, 603–621 CrossRef CAS; J. Fraga-Dubreuil and M. Poliakoff, 2nd International Symposium on Green and Sustainable Chemistry, Delhi, India, 2006 Search PubMed.
- M. D. Bermejo and M. J. Cocero, AIChE J., 2006, 52, 3933–3951 CrossRef CAS; G. Brunner, J. Supercrit. Fluids, 2009, 47, 382–390 Search PubMed.
- C. Aymonier, A. Loppinet-Serani, H. Reveron, Y. Garrabos and F. Cansell, J. Supercrit. Fluids, 2006, 38, 242–251 CrossRef; A. Cabanas and M. Poliakoff, J. Mater. Chem., 2001, 11, 1408–1416 RSC.
- H. Weingartner and E. U. Franck, Angew. Chem., Int. Ed., 2005, 44, 2672–2692 CrossRef.
- L. M. Dudd, E. Venardou, E. Garcia-Verdugo, P. Licence, A. J. Blake, C. Wilson and M. Poliakoff, Green Chem., 2003, 5, 187–192 RSC.
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef.
- P. M. Forster, A. R. Burbank, C Livage, G. Férey and A. K. Cheetham, Chem. Commun., 2004, 368–369 RSC; P. M. Forster and A. K. Cheetham, Microporous Mesoporous Mater., 2004, 73, 57–64 CrossRef CAS.
- J. Liang and G. K. H. Shimizu, Inorg. Chem., 2007, 46, 10449–10451 CrossRef CAS . See also: J-S. Hu, Y-J. Shang, X-Q. Yao, Y-Z. Li, Z-J. Guo, H-G. Zheng and Z-L. Xue, Cryst. Growth Des., 2010, 10, 2676–2684 Search PubMed.
- O. K. Farha, K. L. Mulfort and J. T. Hupp, Inorg. Chem., 2008, 47, 10223–10225 CrossRef CAS.
- X. Zhao, B. Xiao, A. J. Fletcher, K. M. Thomas, D. Bradshaw and M. J. Rosseinsky, Science, 2004, 306, 1012–1015 CrossRef CAS; H. J. Choi, M. Dincă and J. R. Long, J. Am. Chem. Soc., 2008, 130, 7848–7850 CrossRef CAS; S. Yang, X. Lin, A. J. Blake, G. S. Walker, P. Hubberstey, N. R. Champness and M. Schröder, Nat. Chem., 2009, 1, 487–493 CrossRef CAS.
- L. Czepirski and J. Jagiello, Chem. Eng. Sci., 1989, 44, 797–801 CrossRef CAS; X. Zhao, S. Villar-Rodil, A. J. Fletcher and K. M. Thomas, J. Phys. Chem. B, 2006, 110, 9947–9955 CrossRef CAS; X. Zhao, B. Xiao, A. J. Fletcher and K. M. Thomas, J. Phys. Chem. B, 2005, 109, 8880–8888 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Pore size distribution analysis, derivation of the isosteric heats of adsorption, reaction of Zn(NO3)2 with (H4L) at two different temperatures. CCDC reference number 831133. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1gc15726d |
| ‡ Crystal data: [Zn2(C34O8H18)]·3(H2O), M = 369.64, monoclinic, space groupC2/c, a = 31.97 (4), b = 6.334 (9), c = 20.49(3) Å, V = 3590 (9), Z = 8, T = 150 (2)K, μ = 1.402 mm−1, 5442 reflections measured, 2801 unique (Rint = 0.102) which were using in all calculations. Final R1 = 0.137 [1302 F > 4σ(F)], wR2[all F2] = 0.409, maximum ΔF peak 1.16 eÅ−3. |
| This journal is © The Royal Society of Chemistry 2012 |