Catalysis in flow: Au-catalysed alkylation of amines by alcohols†
Natalia
Zotova
a,
Felicity J.
Roberts
b,
Geoffrey H.
Kelsall
b,
Alan S.
Jessiman
c,
Klaus
Hellgardt
*b and
King Kuok (Mimi)
Hii
*a
aDepartment of Chemistry, Imperial College London, Exhibition Road, South Kensington, London, SW7 2AZ, United Kingdom. E-mail: mimi.hii@imperial.ac.uk
bDepartment of Chemical Engineering and Chemical Technology, Imperial College London, Exhibition Road, South Kensington, London, SW7 2AZ, United Kingdom. E-mail: k.hellgardt@imperial.ac.uk
cPfizer Global Research & Development, Ramsgate Road, Sandwich, CT13 9NJ, United Kingdom
First published on 24th November 2011
Abstract
By using a greater reaction space afforded by a flow reactor, commercially available Au/TiO2 can be used for highly selective direct alkylation of amines by alcohols, without the need for an inert atmosphere or base. A brief survey of substrates includes the alkylation of aromatic, aliphatic and chiral amines by a number of primary and secondary alcohols, in high yield and selectivity. The synthesis of Piribedil, a drug used in the treatment of Parkinson's disease, can be achieved in a single synthetic operation without the need for column chromatography. Mechanistic aspects of the reaction were revealed through modelling of reaction profiles, and the origin of selectivity is attributed to the accessibility of high temperature. The presence of water was found to be crucial for catalyst activity.
1 Introduction
The ability to construct increasingly complex molecular structures is a key challenge for organic synthesis, fuelling the demand for synthetic methodologies that can deliver considerable step-economy and high efficiency.1,2 Concurrently, the overall sustainability of a chemical process is important for it to be adopted on an industrial scale. This involves issues such as atom-economy, E-factors and selectivity, which ultimately governs economical and environmental costs of chemical production.3Alcohol activation for nucleophilic substitution is frequently used for the preparation of fine chemicals and active pharmaceutical ingredients (APIs), where there is an urgent need for better methodologies. The direct nucleophilic substitution of an alcohol does not occur easily, and generally requires replacement of the OH by a better leaving group, e.g.halides or OSO2R. In the pharmaceutical industry, it has been estimated that 64% of all nitrogen substitution reactions are alkylations.4 These reactions are very often difficult to control, due to significant side reactions (over-alkylation or competitive elimination), as well as practical limitations, particularly safety and disposal issues associated with the use of mutagenic alkylating reagents.
An alternate strategy is to employ a catalyst to oxidise an alcohol to a reactive carbonyl compound, which condenses with the amine to form an imine, before it is reduced to the amine (Scheme 1). The redox steps can be achieved using a catalyst to transfer H2 from the alcohol to the iminevia metal-hydride intermediates (‘borrowing hydrogen’).5 Perhaps unsurprisingly, metal complexes that are also known to effect reversible dehydrogenation of alcohols (transfer hydrogenation catalysts) have been studied extensively in this context. Accordingly, homogeneous Ru and Ir catalysts are the most prevalent and effective.5,6 Some Cu and Fe catalysts have been reported, however, up to 1 equivalent of base is required.7–9 In terms of practicality, heterogeneous catalysts are much more attractive, as they can be easily separated from the product stream. In this regard, Ru(OH)x/TiO2 was found to have the widest applicability, including the preparation of highly substituted amines from urea,10,11 and the alkylation of (hetero)aromatic amines.12 The scope of first-row transition metal catalysts (Cu, Fe) appears to be limited to certain substrates, such as activated benzyl alcohols or aromatic amines.13–15 More recently, a heterogeneous Ag/Mo oxide catalyst has been shown to be effective for the alkylation of a number of aromatic amines, carboxamides and sulfonamides. However, the system required 20–40 mol% of base (t-BuOK for the alkylation of amines), hence requiring column chromatography for product purification.16
 | ||
| Scheme 1 Direct alkylation of an amine by an alcohol by sequential redox processes. | ||
Heterogeneous gold catalysts are known to be highly effective for the selective oxidation of alcohols17 as well as the hydrogenation of aldehydes, ketones and imines.18 Thus, there have been several attempts to utilise heterogeneous gold catalysts for the direct alkylation of amines by alcohols. Using benzyl alcohol (1a) and aniline (1b) as typical model substrates (Scheme 2), initial studies largely produced the intermediate imine 3 as a major product (Table 1, entries 1 and 2), which was produced exclusively if the reaction was performed under an O2 atmosphere (entries 3 and 4). More recently, the catalytic activity of Au/TiO2 was re-examined under 5 atm of N2 in an autoclave. In this case, very high selectivity was achieved only by using Au catalyst of very small particle size (entry 5 vs. entry 6).
| Entry | [Au] |
1a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2a, conditions :2a, conditions |
Conv.a (%) |
3:4a |
Ref. |
|---|---|---|---|---|---|
| a GC conversions based on alcohol. b Product mixture contained PhCHO and PhCO2Bn side products. c Product mixture contained benzene and toluene side products. d HDP: Hydroxyapatite. e Mean particle size ∼1.8 nm. f <0.5% ‘others’. g Obtained from WGC, mean particle size, ∼3.2 nm. h 1% ‘others’. | |||||
| 1 | Au/TiO2 | 1:1, Cs2CO3, toluene, N2, 110 °C |
>99 | 22:15b | 19 |
| 2 | Au/MgO | 1:3, PhCF3, N2, 180 °C |
93 | 49:38c |
20 |
| 3 | Au/HDPd | 1:1, O2, toluene, 60 °C |
99 | >99:1 |
21 |
| 4 | Au/TiO2 | 1:1, O2, CH3OK (10 mol%), CH3OH, r.t. |
7 | 99:1 |
22 |
| 5 | Au/TiO2-VSe | 1:1, 5 atm. N2, 120 °C, toluene |
>99 | 7.5:92f |
23 |
| 6 | Au/TiO2g | 1:1, 5 atm. N2, 120 °C, toluene |
71 | 6:64h |
23 |
 | ||
| Scheme 2 Model reaction: alkylation of aniline by benzyl alcohol. | ||
2 Results and discussion
Description of the flow system
Previously, we have demonstrated the advantages of using a continuous flow reactor for the catalytic oxidation of alcohols by O2.24 Using a small packed bed of heterogeneous catalyst (70 × 4 mm), reactions can be performed safely at elevated temperature and pressures, while precise control of reaction parameters ensures reproducibility and a high space–time yield. Herein, we will demonstrate a successful application of the flow system to gold-catalysed alkylation of amines by alcohols, which can be achieved under highly practical and atom-economical conditions.Using commercially available Au/TiO2 as catalyst, the reaction between model substrates 1a and 2a was examined under continuous recycle mode conditions (Fig. 1), where a mixture of the substrates in toluene was pressurised and passed through a heated cartridge containing 1 wt% Au/TiO2 in a packed bed. The mixture of substrates and product(s) was collected in a reservoir and then re-circulated through the system. By separating the bulk of reactants from the catalyst, the system can be rendered inherently safe by containing the reaction zone, operating under high temperatures and pressures, within a small reactor volume (< 1 mL). The system also allows for easy access to the reaction mixture for reaction progress analysis using a range of online and offline tools.
 | ||
| Fig. 1 Configuration of the flow reactor (continuous recycle mode). | ||
Effect of temperature
Applying a pressure of 50 bar to maintain a single-phase flow, a marked improvement in conversion and selectivity for 4a was observed when increasing the reaction temperature from 130 through 150 to 180 °C (Table 2, entries 1–3). Furthermore, by adopting nearly equimolar equivalents of the reactants at a higher concentration and lower catalytic loading, the reaction proceeded smoothly to completion with extremely high selectivity for the formation of the secondary amine 4a – with less than 3% of by-product being formed (entry 4).| Entry | T/°C |
1a:2a/M |
[Au] (mol%) | t/h | Conv.b (%) |
3:4a:PhCHO |
|---|---|---|---|---|---|---|
| a Reaction conditions: a mixture of 1a, 2a and dodecane (0.35 mL, internal standard) in toluene (10 mL) was circulated through a heated cartridge of 1.5 wt% Au/TiO2, at a flow rate of 1.5 mL min−1 (residence time = 27 s), 50 bar pressure. b GC conversions based on aniline. | ||||||
| 1 | 130 | 0.29:0.16 |
2.7 | 7 | 65 | 27:65:8 |
| 2 | 150 | 0.29:0.16 |
2.7 | 4 | >99 | 0:94:6 |
| 3 | 180 | 0.16:0.16 |
2.7 | 1 | >99 | 0:93:7 |
| 4 | 180 | 0.51:0.5 |
0.9 | 3 | 99 | <2:97:<1 |
As a comparison, the same reaction mixture and the Au/TiO2 catalyst was subjected to reflux under an N2 atmosphere at ambient pressure. Under these conditions, only 35% conversion of the aniline, and 68% selectivity for 4a can be achieved, broadly in line with the earlier report (Table 1, entry 6).
Hence, by expanding the reaction space, the effectiveness of a gold catalyst can be significantly enhanced, in terms of turnover and selectivity. Based on the result of the last entry of Table 2 a turnover frequency (TOF) of ca. 37 h−1 can be derived, which is broadly compatible, if not better, than other reported homo- and heterogeneous catalytic systems. The efficiency of the system is also accompanied by its practicality – high selectivity for the amine can be obtained with the reaction mixture left exposed to air over the course of the reaction, i.e. the need for rigorously anaerobic conditions, required by all the previous catalytic systems, is not necessary in this system.
Substrate scope
The potential application of the catalytic flow system is demonstrated by conducting a small substrate screen with a selection of primary and secondary alcohols and amines (Table 3). Electron-rich and electron-poor substituents on benzyl alcohol (entries 1–3) and aniline (entries 4 and 5) can be introduced, with very high conversions achieved in just a few hours. The reaction of benzyl alcohol and optically pure α-methylbenzylamine (2c) proceeded very smoothly to give the substituted product 4f with complete retention of stereochemistry (entry 6).|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | R1, R2 | R3 | Product (4) | x(mol%) | T/°C | t/h | Conv.b (%) | Selectivityc (%) |
| a Reaction conditions: amine (0.5 M), alcohol (0.5 M), 0.9–1.8 mol% Au, toluene (10 mL), 50 bar, 1.5 mL min−1 (τ = 27 s). b Determined by GC, using dodecane as internal standard. c Determined by 1H NMR. d Product is enantiomerically pure, as determined by chiral HPLC. | ||||||||
| 1 | Ph, H (1a) | Ph (2a) | 4a | 0.9 | 180 | 3 | 99 | 97 |
| 2 | 4-anisyl, H (1b) | 2a |

|
0.9 | 180 | 1 | 93 | 93 |
| 3 | 4-ClC6H4, H (1c) | 2a |

|
1.8 | 180 | 2 | 83 | 93 |
| 4 | 1a | 2-MeC6H4 (2b) |

|
1.8 | 180 | 2 | 99 | 97 |
| 5 | 1a | 4-ClC6H4 (2c) |
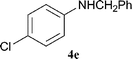
|
1.8 | 180 | 2 | 97 | 94 |
| 6d | 1a | (S)-Ph(Me)CH (2d) |

|
1.8 | 180 | 7 | 91 | 98 |
| 7 | PhCH2, H (1d) | 2a |
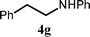
|
1.8 | 200 | 7 | 59 | >99 |
| 8 | 1a | -(CH2)5- (2e) |

|
1.8 | 200 | 6 | 98 | 100 |
| 9 | -(CH2)5- (1e) | 2a |

|
1.8 | 200 | 7 | 85 | 100 |
| 10 | Ph, Me (1f) | n-C6H13 (2f) |

|
1.8 | 200 | 7 | 85 | >99 |

For less activated substrates (entries 7–10), good conversions can be obtained by increasing the temperature to 200 °C; excellent selectivities can be achieved with an aliphatic alcohol (entry 7), a secondary amine (entry 8), as well as cyclic and acyclic secondary alcohols (entries 9 and 10). ICP analyses of crude reaction mixtures did not reveal any detectable metal content (<0.02 ppm), thus catalyst leaching does not occur under these conditions.
The synthetic utility of the system is also demonstrated for more complex substrates by the synthesis of Piribedil 4k, a piperazine-pyrimidine derivative used in the treatment of Parkinson's disease.25 A solution of piperonyl alcohol 1g and 1-(pyrimidin-2-yl)piperazine 2g in toluene was circulated through the heated catalyst maintained at 200 °C and 50 bar (Scheme 3). After 24 h, the reaction mixture was simply evaporated and the residue recrystallised from ethanol–water to furnish the desired product in 77% yield with high purity, without the need for column chromatography.
 | ||
| Scheme 3 Synthesis of Piribedil 4k by direct alkylation of piperazine-pyrimidine derivative 2g by 1g. | ||
Reaction mechanism
The reaction between benzyl alcohol and aniline was analysed in our preliminary kinetic/mechanistic study. Remarkably, the selectivity and rate of the reaction are unaffected by applying pressures of between 5–100 bar (Fig. S4, ESI†). This implies that nascent H2 is not present in the system, i.e.reduction of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond occurs exclusively through adsorbed hydrides. In contrast, reaction temperature has a dramatic effect on the selectivity of the reaction: a significant accumulation of imine and benzaldehyde was observed at lower temperature (130 °C), which was suppressed when the reaction temperature was raised to 150 or 180 °C (Fig. 2a, b and c).
N bond occurs exclusively through adsorbed hydrides. In contrast, reaction temperature has a dramatic effect on the selectivity of the reaction: a significant accumulation of imine and benzaldehyde was observed at lower temperature (130 °C), which was suppressed when the reaction temperature was raised to 150 or 180 °C (Fig. 2a, b and c).
A closer examination of the reaction profile revealed that the presence of imine 3 was observed in the initial stages of all three reactions. At the lower reaction temperature (130 °C), the concentration of oxidised products (imine and aldehyde) continues to rise over the course of the reaction. In contrast, at higher temperatures, the formation of the imine ceased to be competitive, and started to behave as a reaction intermediate (Fig. 2d).
![a), b) and c) Temporal reaction profiles between 1a and 2a corresponding to entries in Table 2 ( = [2a], = [3], ▲ = [4a], = [PhCHO]); and d) effect of temperature on the temporal evolution of imine 3.](/image/article/2012/GC/c1gc16118k/c1gc16118k-f2.gif) | ||
Fig. 2 a), b) and c) Temporal reaction profiles between 1a and 2a corresponding to entries in Table 2 ( = [2a], = [2a],  = [3], ▲ = [4a], = [3], ▲ = [4a],  = [PhCHO]); and d) effect of temperature on the temporal evolution of imine 3. = [PhCHO]); and d) effect of temperature on the temporal evolution of imine 3. | ||
A catalytic cycle was thus proposed based on these observations, from which four rate equations can be derived. The competitive processes leading to the formation of the imine by-product is represented by two equilibria KA and KB, associated with the desorption of the aldehyde from the catalyst surface, and the subsequent condensation reaction, respectively (Scheme 4). The reaction progress curves obtained under different reaction conditions (Fig. 2a, b and c) were subjected to kinetic analysis, using the model developed (in Berkeley Madonna26), and the results are summarised in Table 4.
| T/°C | k 1/s−1 | K A·KB | k 2/M−1 s−1 |
|---|---|---|---|
| a See ESI.† | |||
| 130 | 2.05 × 10−5 | 6.96 × 10−2 | 3.58 × 10−2 |
| 150 | 8.61 × 10−5 | 3.78 × 10−1 | 0.1 |
| 180 | 4.53 × 10−4 | 1.46 | 5.65 × 10−2 |
| E a/kJ mol−1 | 93.7 | 91.2 | — |
 | ||
| Scheme 4 Proposed catalytic cycle and associated rate equations. | ||
The conversion of benzyl alcohol to benzaldehyde catalysed by Au/CeO2 had been extensively studied previously in a body of work by Corma and co-workers,27 whereby benzyl alcohol undergoes fast and reversible binding to the catalyst surface to give an alkoxide species (I). In the present system, Langmuir–Hinshelwood type saturation kinetics was also observed, where the rate-limiting step is found to be the formation of the benzaldehyde (k1), with an activation energy of 93.7 kJ mol−1. In all three cases, the initial oxidation of the alcohol to the carbonyl compound is the rate-limiting step of the catalytic cycle.
Slow β-hydride elimination occurs to generate II, consisting of gold-hydride and surface-bound benzaldehyde, which may be liberated into solution. Condensation with aniline gives adsorbed or free imines III or 3, respectively. In our proposed model, only adsorbed imine III can be reduced by AuH to the desired product 4a. The rate of the hydrogenation step (k2) is found to be at least two orders of magnitude faster than the dehydrogenation of the alcohol, and is likely to be irreversible. This is supported by the observation that the stereochemistry of a chiral amine substrate can be retained during the process (Table 2, entry 6).
The selectivity of the reaction is defined by the relative ratio of amine (4a) to imine (3), formed through the partitioning of reaction pathway via the common intermediate II. This is dependent upon the overall rate of conversion of II to 4a (k2), and the rate of formation of imine 3 from the desorbed benzaldehyde, expressed by a combination of two equilibrium constants (KA·KB). At 130 °C, k2 is sufficiently slow, such that the favourable formation of the imine (KA·KB = 6.96 × 10−2) is competitive. At higher temperatures, imine formation becomes disfavoured, particularly at higher conversions (where the amount of water in the system increases). This effectively counteracts the desorption process by increasing the amount of benzaldehyde in the system, thus allowing it to re-enter the productive catalytic cycle. At 180 °C, very high selectivity can be achieved, even though the process is dominated by the larger KA·KB equilibria. It is interesting that k2 appears to decrease when the temperature was raised from 150 to 180 °C. It is important to keep in mind that k2 embraces several key bond-forming steps (formation of imine, reduction and liberation from the surface), and this break from the Arrhenius linearity suggests there may be a change in the mechanism of at least one of these steps.
(Unexpected) Effect of water
Given that water is generated as a by-product of this process, it is not unreasonable to assume that the removal of water can have a beneficial effect on the process. With this in mind, further experiments were conducted under anhydrous conditions, with a sample of a catalyst dried in an oven overnight at 150 °C, and by passing the reaction stream through a cartridge of molecular sieves. Rather surprisingly, catalyst activity and selectivity were practically annihilated under these conditions (Fig. 3) – only 10% conversion was achieved after 3 h with 29% selectivity for the amine product. It is notable that even the formation of benzaldehyde was sequestered under these conditions, i.e. water plays a crucial role in activating the catalyst at the onset of the reaction. The interaction between water and TiO2 is key to several important energy and environmental applications (e.g. solar-hydrogen production and photodecomposition of organic pollutants). As the subject of numerous studies over the last few decades,28 it is generally accepted that water can dissociate over certain TiO2 surfaces to generate –OH and –H species which affects its acid–base properties. Very recently, the interaction of alcohols with TiO2 surfaces has also been investigated at the solid–liquid boundary.29 Hence, we postulated that the presence of water is critical in activating the surface of TiO2 towards adsorption of substrates that are critical for catalytic activity.![Reaction between 1a and 2a under anhydrous conditions ( = [2a], = [3], ▲ = [4a], = [PhCHO]). Reaction conditions: 180 °C, 50 bar, 1.5 mL min−1, 1 mol% Au/TiO2.](/image/article/2012/GC/c1gc16118k/c1gc16118k-f3.gif) | ||
Fig. 3 Reaction between 1a and 2a under anhydrous conditions ( = [2a], = [2a],  = [3], ▲ = [4a], = [3], ▲ = [4a],  = [PhCHO]). Reaction conditions: 180 °C, 50 bar, 1.5 mL min−1, 1 mol% Au/TiO2. = [PhCHO]). Reaction conditions: 180 °C, 50 bar, 1.5 mL min−1, 1 mol% Au/TiO2. | ||
3 Conclusions
The use of gold as a viable heterogeneous catalyst for the alkylation of amines by alcohols has been achieved for the first time under aerobic conditions using a continuous flow reactor and commercially-available catalysts that are otherwise not selective in batch reactors.30 Very high reactivity and selectivity can be accomplished in several systems.Compared to previously reported systems, the gold-catalysed flow process has a number of notable advantages:
(i) Catalysis occurs entirely at the surface of Au–TiO2, and there is no detectable leaching;
(ii) Unlike all other catalysts, rigorous exclusion of air and/or moisture is not necessary;
(iii) Reactions can be performed using commercially-available catalysts from several sources;
(iv) Equimolar amounts of the reactants can be used, and addition of exogenous base is not necessary, thus greatly simplifying product purification; and
(v) The system allows for easier and safer scale-up of processes.
Key steps of the catalytic cycle have been elucidated by kinetic modelling. The study show that the inherent lower catalytic activity of the gold catalyst can be overcome by adopting a higher reaction temperature (>150 °C) afforded by a flow system, where the formation of imine become reversible and can re-enter the productive catalytic cycle.
The current study also uncovered a possible change in reaction mechanism at 180 °C, and an unexpected role of water in the activation of the catalyst. These will be investigated further. Modifications of the catalyst will also be attempted to further improve its efficiency, including widening of the reaction scope to a broader range of substrates.
4 Experimental
General
Starting materials were procured commercially and used without purification. Reactions were performed in reagent grade toluene. The conversion of alcohols to products was monitored using a HP6890 gas chromatography system fitted with an SPB-5 column (30 m × 0.2 μm × 0.8 μm). The percentage of conversion/selectivity was determined by comparison with known standards, using a calibration plot for product and reactant, against dodecane as an internal standard. The identity of all products was further verified by comparison of their 1H NMR spectra to that reported, recorded in CDCl3 using a Bruker Avance NMR spectrometer at 400 MHz. ICP analyses (%Au) were performed using a Perkin–Elmer 2000 DV ICP-OE spectrometer. The BET surface area and mean pore size of the catalyst were obtained by low temperature N2 adsorption studies at 77 K using a Micromeritics Tristar 3000 instrument.Catalyst characterisation
1 wt% Au/TiO2 obtained from Strem (AUROlite™ Au/TiO2), was ground using a mortar and pestle and sieved. Particle sizes of 125–250 microns were employed. Average gold crystallite size is ca. 2–3 nm. BET surface area = 40 m2 g−1, mean pore size = 0.26 cm3 g−1, mean pore diameter = 26 nm.Catalytic reactions
Reactions were performed using a ThalesNano X-Cube™ flow reactor.31 A solution of the alcohol (0.29/0.5 M) and amine (0.16/0.5 M) in toluene (10 mL) containing dodecane (0.18 M) as an internal standard, was (re)circulated through a heated cartridge containing 1 wt% Au/TiO2 (0.6–1.8 g) at 180–200 °C, under an applied pressure of between 5–100 bar. The progress of the reaction was monitored by GC analysis. Upon completion, the solution was evaporated to afford the corresponding substituted amine with >90% recovery. The purity and identity of the product were verified by 1H NMR spectroscopy (ESI†).Synthesis of Piribedil (4k)
A mixture of the alcohol 1g (760 mg, 5 mmol) and piperazine 2g (827.4 mg, 5 mmol) in toluene (10 mL) was circulated for 24 h through 1.8 mol% of Au/TiO2 mounted in 2 heated cartridges, held at 200 °C and 50 bar pressure. The reaction mixture was evaporated to dryness, and the residue was recrystallised from hot EtOH–H2O, to give the desired product as a buff-coloured solid (1.15 g, 77%). 1H NMR data (ESI†) is in accordance with reported values.32Acknowledgements
The work is funded by an EPSRC grant for Flow Chemistry (EP/G027544/1), with additional support by Pfizer Inc. We thank the Pharmacat Consortium for useful discussions.Notes and references
- P. A. Wender, V. A. Verma, T. J. Paxton and T. H. Pillow, Acc. Chem. Res., 2008, 41, 40–49 CrossRef CAS.
- T. Newhouse, P. S. Baran and R. W. Hoffmann, Chem. Soc. Rev., 2009, 38, 3010–3021 RSC.
- P. J. Dunn, A. S. Wells and M. T. Williams, ed., Green Chemistry in the Pharmaceutical Industry, Wiley-VCH, 2010 Search PubMed.
- D. J. C. Constable, P. J. Dunn, J. D. Hayler, G. R. Humphrey, J. L. Leazer, R. J. Linderman, K. Lorenz, J. Manley, B. A. Pearlman, A. Wells, A. Zaks and T. Y. Zhang, Green Chem., 2007, 9, 411–420 RSC.
- T. D. Nixon, M. K. Whittlesey and J. M. J. Williams, Dalton Trans., 2009, 753–762 RSC.
- G. Guillena, D. J. Ramon and M. Yus, Chem. Rev., 2010, 110, 1611–1641 CrossRef CAS.
- F. Shi, M. K. Tse, X. J. Cui, D. Gordes, D. Michalik, K. Thurow, Y. Q. Deng and M. Beller, Angew. Chem., Int. Ed., 2009, 48, 5912–5915 CrossRef CAS.
- A. Martinez-Asencio, D. J. Ramon and M. Yus, Tetrahedron Lett., 2010, 51, 325–327 CrossRef CAS.
- Y. Zhao, S. W. Foo and S. Saito, Angew. Chem., Int. Ed., 2011, 50, 3006–3009 CrossRef CAS.
- J. L. He, J. W. Kim, K. Yamaguchi and N. Mizuno, Angew. Chem., Int. Ed., 2009, 48, 9888–9891 CrossRef CAS.
- K. Yamaguchi, J. L. He, T. Oishi and N. Mizuno, Chem.–Eur. J., 2010, 16, 7199–7207 CrossRef CAS.
- J. W. Kim, K. Yamaguchi and N. Mizuno, J. Catal., 2009, 263, 205–208 CrossRef CAS.
- P. R. Likhar, R. Arundhathi, M. L. Kantam and P. S. Prathima, Eur. J. Org. Chem., 2009, 5383–5389 CrossRef CAS.
- R. Martinez, D. J. Ramon and M. Yus, Org. Biomol. Chem., 2009, 7, 2176–2181 CAS.
- J. He, K. Yamaguchi and N. Mizuno, Chem. Lett., 2010, 39, 1182–1183 CrossRef CAS.
- X. Cui, Y. Zhang, F. Shi and Y. Deng, Chem.–Eur. J., 2011, 17, 1021–1028 CrossRef CAS.
- C. Della Pina, E. Falletta, L. Prati and M. Rossi, Chem. Soc. Rev., 2008, 37, 2077–2095 RSC.
- L. McEwan, M. Julius, S. Roberts and J. C. Q. Fletcher, Gold Bull., 2010, 43, 298–306 CrossRef CAS.
- T. Ishida, N. Kawakita, T. Akita and M. Haruta, Gold Bull., 2009, 42, 267–274 CrossRef CAS.
- A. Corma, T. Rodenas and M. J. Sabater, Chem.–Eur. J., 2010, 16, 254–260 CrossRef CAS.
- H. Sun, F. Z. Su, J. Ni, Y. Cao, H. Y. He and K. N. Fan, Angew. Chem., Int. Ed., 2009, 48, 4390–4393 CrossRef CAS.
- S. Kegnaes, J. Mielby, U. V. Mentzel, C. H. Christensen and A. Riisager, Green Chem., 2010, 12, 1437–1441 RSC.
- L. He, X.-B. Lou, J. Ni, Y.-M. Liu, Y. Cao, H.-Y. He and K.-N. Fan, Chem.–Eur. J., 2010, 16, 13965–13969 CrossRef CAS.
- N. Zotova, K. Hellgardt, G. H. Kelsall, A. S. Jessiman and K. K. Hii, Green Chem., 2010, 12, 2157–2163 RSC.
- M. J. Millan, Pharmacol. Ther., 2010, 128, 229–273 CrossRef CAS.
- http://www.berkeleymadonna.com/ .
- A. Abad, A. Corma and H. García, Chem.–Eur. J., 2008, 14, 212–222 CrossRef CAS.
- C. H. Sun, L. M. Liu, A. Selloni, G. Q. Lu and S. C. Smith, J. Mater. Chem., 2010, 20, 10319–10334 RSC.
- V. M. Sanchez, E. de la Llave and D. A. Scherlis, Langmuir, 2011, 27, 2411–2419 CrossRef CAS.
- Au/TiO2 attained from other sources such as World Gold Council and donated by Johnson Matthey plc, were also found to be effective under these conditions.
- ThalesNano Nanotechnology Inc., Graphisoft Park, H-1031 Budapest Zahony u. 7, Hungary (see: http://www.thalesnano.com/products/x-cube).
- A. J. A. Watson, A. C. Maxwell and J. M. J. Williams, J. Org. Chem., 2011, 76, 2328–2331 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: details of kinetic analysis and fitted curves, pressure effects, Arrhenius plots, and 1H NMR spectra of products. See DOI: 10.1039/c1gc16118k |
| This journal is © The Royal Society of Chemistry 2012 |