Direct synthesis of hydrogen peroxide using Au–Pd-exchanged and supported heteropolyacid catalysts at ambient temperature using water as solvent
Edwin N.
Ntainjua
,
Marco
Piccinini
,
Simon J.
Freakley
,
James C.
Pritchard
,
Jennifer K.
Edwards
,
Albert F.
Carley
and
Graham J.
Hutchings
*
Cardiff University, School of Chemistry, Main Building, Park Place, Cardiff, CF10 3AT, UK
First published on 8th November 2011
Abstract
The direct synthesis of hydrogen peroxide from molecular H2 and O2 represents a green and economic alternative to the current anthraquinone process used for the industrial production of H2O2. In order for the direct process to compete with the anthraquinone process, there is a need for enhanced H2O2 yields and H2 selectivity in the process. We show that Au–Pd-exchanged and supported Cs-containing heteropolyacid catalysts with the Keggin structure are considerably more effective in achieving high H2O2 yields in the absence of acid or halide additives than previously reported catalysts. The Au–Pd-exchanged Cs-heteropolyacid catalysts also show superior H2O2 synthesis activity under challenging conditions (ambient temperature, water-only solvent and CO2-free reaction gas). Au plays a crucial role in achieving the improved performance of these heteropolyacid-based catalysts. The heteropolyacid limits the subsequential hydrogenation/decomposition of H2O2.
Introduction
The direct synthesis of hydrogen peroxide (H2O2) from molecular hydrogen and oxygen represents a potentially economic and environmentally benign alternative to the current industrial process. At present, H2O2 is produced by the sequential hydrogenation and oxidation of an alkyl anthraquinone and over 2 M tonnes per year is manufactured in this way. Most of the H2O2 produced globally is used in bleaching and disinfectant applications, such as in the textile/paper and pulp industry and in medical practice, respectively. Legislative pressure to limit the use of chlorine in bleaching applications means that more H2O2 has to be produced in order to meet the high demand for bleaches. Recent interest in the use of H2O2 as a green oxidant for chemical synthesis has partly accounted for the considerable increase (10% per annum1) in its global production.The current anthraquinone process used for the industrial production of H2O2 requires high capital investment and involves the production and transportation of very concentrated H2O2 solutions; contrarily, a direct synthesis process would allow for small scale production of dilute H2O2 solutions at point of use in a green and inexpensive manner. The direct process therefore eliminates the risk of accidents2 associated with the transportation of such concentrated H2O2 solutions from the production plants to the point of use.
Several research studies have reported the use of supported monometallic Pd3–20 and bimetallic Au–Pd catalysts21–33 for the direct synthesis of H2O2. Although the monometallic Pd catalysts are the most widely studied, Au–Pd bimetallic catalysts have been shown to be significantly more effective. TiO2- and carbon-supported Au–Pd catalysts constitute the most effective28–30 supported bimetallic Au–Pd catalysts for the direct synthesis of H2O2. The carbon-based Au–Pd catalyst is the most effective, yielding significantly higher H2O2 productivity and H2 selectivity compared to the monometallic Pd and other supported Au–Pd catalysts. Although we consider that the discovery of these highly active Au–Pd catalysts does represent a significant advance for the direct process; further advances are needed. To compete with the indirect anthraquinone process, there is a need to optimise the H2O2 yield and H2 selectivity in the direct synthesis process. This goal can be achieved by the formulation of more effective catalysts and by tuning reaction conditions so as to achieve the optimum catalytic performance. Most of the Pd and Au–Pd supported catalysts used to date for the direct synthesis of H2O2 are also active for the sequential hydrogenation/decomposition of H2O2 to form H2O and/or the hydrogenation of O2 to H2O, thus limiting the amount of H2O2 that can be produced using these catalysts. This represents a prime setback for the direct process and, as such, has constituted the subject of various industrial6–8 and scientific11–20,31–33 studies. In order to limit the undesired side reactions that lead to low H2O2 yields over the monometallic Pd catalysts, acids and halides have to be added to the reaction mixture.10–19Halide incorporation into Pd catalysts has also been employed to suppress H2O2 hydrogenation/decomposition.15–17,20 However, we have recently shown31,32 that the addition of acid and halide to the reaction mixture or the incorporation of halide into the catalyst is not required when using the most effective Au–Pd bimetallic catalyst (Au–Pd/C). Acid and halide addition or halide incorporation only lead to a subtle positive effect at low concentrations. Moreover, the effect of halide addition/incorporation is not sustained upon catalyst re-use as the presence of bromide leads to the redistribution of the Au–Pd nanoparticles and, as a result, induces Au leaching from the catalyst. We have previously reported33 a green and cost-effective method of switching-off the undesired hydrogenation/decomposition of H2O2 during its synthesis over the highly effective Au–Pd/C catalyst. Acid pre-treatment of the carbon support prior to co-impregnation with Au and Pd switches off H2O2 hydrogenation/decomposition and significantly enhances H2O2 productivity. This has prompted us to consider acidic supports as a basis for improved catalyst design. Sun et al.34 and Park et al.35 have recently reported the use of Pd-only insoluble heteropolyacid catalysts, comprising the Keggin structure, for the direct synthesis of H2O2 in the absence of any acid and/or halide additives. Sun et al.34 showed that Pd-based supported heteropolyacid catalysts showed higher H2O2 productivity and selectivity compared to other conventional Pd-only catalysts. Park et al.35 studied Pd-exchanged heteropolyacids (Pd0.15CsxH2.7−xPW12O40) with varying Cs content (2.0–2.7) for H2O2 synthesis and found that the most acidic catalyst (Pd0.15Cs2.5H0.2PW12O40) showed the best performance. Building on these earlier studies we have investigated the effect of substituting some of the Pd present in Pd0.15Cs2.5H0.2PW12O40 with Au and in this paper we report our findings, showing that the addition of Au markedly enhances the activity. We also study the effect of using heteropolyacids with the Keggin structure as supports for Pd, Au and Au–Pd catalysts prepared by impregnation and in this paper we present the results for their use as catalysts for the direct synthesis of hydrogen peroxide.
Experimental
Catalyst preparation
A Pd-exchanged heteropolyacid catalyst (Pd0.15Cs2.5H0.2PW12O40) was prepared using an ion-exchange method. Appropriate amounts of Pd(NO3)2 (Aldrich) and CsNO3 (Aldrich) were dissolved separately in deionised water and added dropwise with stirring to an aqueous solution of H3PW12O40 (Aldrich, 99.999%) containing the appropriate amount of the acid. The resulting solution was continuously stirred while heating at 60 °C for 12 h to obtain a solid product, which was then dried overnight at 70 °C, followed by calcination at 300 °C for 2 h to form the final catalyst. A Pd-free heteropolyacid (Cs2.8H0.2PW12O40) was prepared in a similar manner and used as a support material for the preparation of supported Pd, Au and Au–Pd catalysts. Au–Pd-exchanged heteropolyacids (Pd0.1Au0.0333Cs2.5H0.2PW12O40 and Pd0.075Au0.05Cs2.5H0.2PW12O40) were also prepared using a similar ion exchange method. In this case, aqueous solutions of HAuCl4·3H2O and Pd(NO3)2 were added drop-wise to a solution of H3PW12O40 followed by drop-wise addition of dissolved CsNO3 after stirring of the mixture for 5 min.Supported Au–Pd catalysts comprising 2.5 wt% Au/2.5 wt% Pd/support were prepared using the following standard co-impregnation method (all quantities stated are per g of finished catalyst). PdCl2 (0.042 g, Johnson Matthey) was added to aqueous HAuCl4·3H2O solution (2.5 ml, 5 g in 250 ml) and stirred at 80 °C until the Pd dissolved completely. The appropriate support (0.95 g; carbon (G60, Aldrich, acid-treated and untreated) or Cs2.8H0.2PW12O40) was then added to the solution and stirred to form a paste. The paste was dried (110 °C, 16 h) before calcination (400 °C, 3 h for the carbon catalysts and 300 °C, 2 h for the heteropolyacid-based catalysts). Monometallic Au and Pd supported heteropolyacid catalysts were also prepared using the same impregnation method.
Catalyst characterisation and testing
Hydrogen peroxide synthesis and hydrogenation was evaluated using a Parr Instruments stainless steel autoclave with a nominal volume of 100 ml and a maximum working pressure of 14 MPa. To evaluate each catalyst for H2O2 synthesis the following standard reaction conditions were used. The autoclave was charged with catalyst (0.01 g) and 8.5 g solvent (5.6 g MeOH and 2.9 g H2O). The charged autoclave was then purged three times with 5% H2/CO2 (0.7 MPa) before filling with 5% H2/CO2 to a pressure of 2.9 MPa at 20 °C. The pressure was allowed to drop to 2.6 MPa as the gas dissolved in the solvent at 20 °C. This was followed by the addition of 25% O2/CO2 (1.1 MPa). The temperature was then allowed to decrease to 2 °C followed by stirring (at 1200 rpm) of the reaction mixture for 30 mins. The above reaction parameters represent the optimum conditions we have previously used for the synthesis of H2O2.22–33 In this study, we systematically varied these reaction conditions so as to investigated the use of ambient temperature, H2O-only solvent and 2%H2/air for the direct synthesis of H2O2. H2O2 productivity was determined by titrating aliquots of the final solution after reaction with acidified Ce(SO4)2 (0.01 M) in the presence of two drops of ferroin indicator.H2O2 hydrogenation experiments were carried out in a similar manner as H2O2 synthesis experiments but without adding the 1.1 MPa 25%O2/CO2. Furthermore, 0.68 g of H2O from the 8.5 g of solvent was replaced by a 50% H2O2 solution to give a reaction solvent containing 4 wt% H2O2. The standard reaction conditions for H2O2 hydrogenation included: 0.01 g catalyst, 8.5 g solvent (5.6 g MeOH, 2.22 g H2O and 0.68 g H2O2 (50%)), 2.9 MPa 5%H2/CO2, 2 °C, 1200 rpm, 30 mins. Just like for H2O2 synthesis, the standard conditions were varied in some experiments to study H2O2 hydrogenation at ambient temperature and when using H2O-only solvent.
FT-IR spectra were measured using a Varian Excalibur 400 FT-IR spectrometer with an UMA 600 microscope. The images were recorded in reflection mode. A small portion of sample was pressed onto a single germanium crystal/microscope base and irradiated by an infra-red source. The sample was exposed to the atmosphere during analysis and so a background spectrum was processed beforehand, allowing subtraction of CO2 peaks situated at: 1300–2000 cm−1 (broad), 2300–2400 cm−1 (sharp), 2840–3000 cm−1 (weak) and 3500–4000 cm−1 (broad).
Laser Raman spectroscopy was performed using a RENISHAW inVia Raman microscope, using a 25mW power laser set at a reflection wavelength of 514 nm. The laser was set to a power output of 5% in order to avoid damaging samples in the study and obtaining the best result clarity.
Investigation of the bulk structure of the materials was carried out using powder X-ray diffraction (XRD) on a (θ–θ) PANalytical X'pert Pro powder diffractometer using a Cu-Kα radiation source operating at 40 KeV and 40 mA. Standard analysis was performed using a 40 min scan between 2θ values of 10–80° with the samples supported on an amorphous silicon wafer. Diffraction patterns of phases were identified using the ICDD data base.
Temperature programmed desorption (TPD) profiles were recorded using a Thermo 1100 series TPDRO. 0.1 g of sample was packed into the sample tube using quartz wool and a volume reducer. The sample was then pre-treated under He while being heated from room temperature to 110 °C at 5 °C min−1, where it was held for 60min. The sample was allowed to cool to room temperature, following this ammonia was passed over the catalyst for 10 min at 20 ml min−1. The samples were then heated from room temperature to 900 °C at 5 °C min−1. The profile was recorded using a TCD with positive polarity and a gain of 10.
XPS measurements were made on a Kratos Axis Ultra DLD spectrometer using monochromatic AlKα radiation (source power 120–180 W). An analyser pass energy of 160 eV was used for survey scans, and 40 eV for detailed acquisition of individual elemental regions. Samples were mounted using double-sided adhesive tape, and binding energies referenced to the C(1 s) binding energy of adventitious carbon contamination taken to be 284.7 eV. Spectra were quantified using CasaXPS (Neil Fairley, UK) and data is presented as surface compositions (atom%).
Results and discussion
Comparison of carbon and heteropolyacid-based Au, Pd and Au–Pd catalysts for H2O2 synthesis and hydrogenation using standard reaction conditions
In our previous studies,22–33 we employed standard conditions specified in Table 1 for H2O2 synthesis and hydrogenation. We have previously shown32 that a Au–Pd/C catalyst prepared by co-impregnation of Au and Pd onto an acid pre-treated carbon support, designated Au–Pd/C(2% HNO3), was the most effective Au–Pd catalyst for the direct synthesis of H2O2 when using these standard reaction conditions. Table 1 compares the rate of H2O2 synthesis and hydrogenation over heteropolyacid catalysts and Au–Pd/C(2% HNO3) using the standard reaction conditions. The Au–Pd supported heteropolyacid catalyst (2.5% Au/2.5% Pd/Cs2.8H0.2PW12O40) was the most active catalyst (for both synthesis and hydrogenation), yielding significantly higher H2O2 productivity than Au–Pd/C(2% HNO3). The Au–Pd/C(2% HNO3) catalyst, however, showed a higher H2O2 synthesis rate than the other heteropolyacid-based catalysts. Since this catalyst does not show any H2O2 hydrogenation/decomposition activity, it is likely to be the most selective catalyst for H2O2 synthesis using standard reaction conditions. Heteropolyacid-supported Pd and Au–Pd catalysts showed higher H2O2 formation activity compared to carbon-supported catalysts with the same Pd or Au–Pd content. Although the heteropolyacid-based Pd and Au–Pd catalysts prepared by ion exchange yield lower H2O2 productivities than the heteropolyacid- and carbon-based supported catalysts, they contain ca. 9–10 times less active metal (Au and/or Pd) present in the supported catalysts. Since the Pd and Au–Pd-exchanged heteropolyacid catalysts yield relatively high H2O2 formation rates (86–96 mol kgcat−1 h−1 compared to 110–198 mol kgcat−1 h−1 for the supported catalysts) irrespective of their very low Pd or Au–Pd content (ca. 0.4–0.6 wt%), we consider them to be promising catalysts for the direct synthesis of H2O2. Fig. 1 shows that the turnover frequency (TOF) for Pd and Au–Pd heteropolyacid catalysts made by ion exchange are ca. 3 times more than for the heteropolyacid- and carbon-supported Au–Pd catalysts. Moreover, these catalysts also show considerably lower H2O2 hydrogenation rates compared to the most active supported catalyst (2.5% Au/2.5% Pd/Cs2.8H0.2PW12O40).| Catalyst | H2O2 Productivitya/mol kgcat−1 h−1 | H2O2 Hydrogenation b/mol kgcat−1 h−1 |
|---|---|---|
| a Rate of hydrogen peroxide production determined using reaction using standard reaction conditions: 5% H2/CO2 (2.9 MPa) and 25% O2/CO2 (1.1 MPa), 8.5 g solvent (5.6 g MeOH + 2.9 g H2O), 0.01 g catalyst, 2 °C, 1200 rpm, 30 min. b Rate of hydrogenation of H2O2 calculated from amount of H2O2 hydrogenated using standard reaction conditions: 2.9 MPa 5%H2/CO2, 8.5 g solvent (5.6 g MeOH, 2.22 g H2O and 0.68 g 50% H2O2), 0.01 g catalyst, 2 °C, 1200 rpm, 30 min. | ||
| Cs2.8H0.2PW12O40 | 1 | 162 |
| Pd0.15Cs2.5H0.2PW12O40 | 96 | 221 |
| Au0.1Cs2.5H0.2PW12O40 | 16 | 347 |
| Pd0.1Au0.0333Cs2.5H0.2PW12O40 | 97 | 384 |
| Pd0.075Au0.05Cs2.5H0.2PW12O40 | 86 | 213 |
| 5% Pd/Cs2.8H0.2PW12O40 | 136 | 281 |
| 5% Au/Cs2.8H0.2PW12O40 | 14 | 103 |
| 2.5% Au/2.5% Pd/Cs2.8H0.2PW12O40 | 198 | 704 |
| 2.5% Au/2.5% Pd/C | 110 | 117 |
| 2.5% Au/2.5% Pd/C (2% HNO3) | 160 | 0 |
![A comparison of TOF for H2O2 synthesis at 2 °C using heteropolyacid-based and carbon-based Au, Pd and Au–Pd catalysts [data corrected to take account of contribution from the Au and Pd-free (heteropolyacid-only) material]. HPA – heteropolyacid, AW – acid washed.](/image/article/2012/GC/c1gc15863e/c1gc15863e-f1.gif) | ||
| Fig. 1 A comparison of TOF for H2O2 synthesis at 2 °C using heteropolyacid-based and carbon-based Au, Pd and Au–Pd catalysts [data corrected to take account of contribution from the Au and Pd-free (heteropolyacid-only) material]. HPA – heteropolyacid, AW – acid washed. | ||
The Au-only exchanged and supported heteropolyacid catalysts showed considerably higher H2O2 productivity compared to Au-only supported catalysts that we have previously reported.29 However, the addition of Au by ion-exchange to the Pd-only heteropolyacid did not significantly alter the catalytic performance under standard reaction conditions. This is in contrast to the synergistic effect observed21–33 for conventional supported Au–Pd catalysts. We have shown that the addition of Au to Pd-only catalysts supported on C, Al2O3, TiO2 and SiO2 lowers H2O2 hydrogenation/decomposition and promotes H2O2 synthesis under standard reaction conditions. Unlike the Au–Pd-exchanged heteropolyacid catalysts, the Au–Pd-supported heteropolyacid catalyst showed higher H2O2 synthesis activity than the Pd-only supported catalyst, thus confirming the promotional effect of Au previously observed for other supported Au–Pd catalysts. This may suggest that the different methods of catalyst preparation (ion-exchange and impregnation) lead to the formation of Au and Pd nanoparticles with varying characteristics (e.g. particle size, location and nature/structure) and as such show different H2O2 synthesis and hydrogenation/decomposition abilities.
The heteropolyacid-only catalyst (Cs2.8H0.2PW12O40) showed the lowest rate of H2O2 formation amongst all catalysts tested. This catalyst was almost inactive for H2O2 synthesis but showed considerable hydrogenation activity. However, the use of this heteropolyacid-only material as a catalyst support for Pd-only and Au–Pd catalysts led to catalysts showing significantly higher H2O2 formation rates than similar Pd-only and Au–Pd catalysts supported on other conventional support materials21–33 (C, Al2O3, TiO2 and SiO2). We have previously shown29 that acidic supports with low isoelectric points (IEP) gave improved catalytic performance for supported-Pd and Au–Pd catalysts than basic supports e.g.MgO. Since the heteropolyacid-based supported catalysts are composed of a more acidic support (with lower IEP) than those in other conventional supported catalysts, we attribute their superior H2O2 synthesis performance under standard conditions to the intrinsically acidic character of the heteropolyacid support.
H2O2 synthesis and hydrogenation at ambient temperature over carbon and heteropolyacid-based Au, Pd and Au–Pd catalysts
H2O2 synthesis is often carried out at sub-ambient temperatures (e.g. 2 °C) in order to avoid subsequent decomposition of H2O2 as it is formed. We have now investigated the effect of carrying out H2O2 synthesis and hydrogenation at ambient temperature (Table 2) while maintaining all other reaction conditions unchanged. The H2O2 hydrogenation rates over all catalysts investigated (except Au0.1Cs2.5H0.2PW12O40 and 5% Au/Cs2.8H0.2PW12O40) were considerably higher at ambient temperature compared to 2 °C. This is not surprising as thermal decomposition of H2O2 is favoured at ambient temperature. Surprisingly, however, Au0.1Cs2.8H0.2PW12O40 and 5% Au/Cs2.8H0.2PW12O40 showed lower H2O2 hydrogenation rates at ambient temperature than at 2 °C. The fact the Au-only heteropolyacid catalyst has a significantly lower H2O2 hydrogenation activity compared to the heteropolyacid-only catalyst demonstrates that the incorporation of Au (by ion-exchange or impregnation) in the heteropolyacid-only material also led to significant decrease in H2O2 hydrogenation at ambient temperature. This suggest that Au inhibits the hydrogenation/decomposition of H2O2 over the heteropolyacid-based catalysts at ambient temperature. This effect is, however, unique for the elevated temperature as we observed the opposite effect (increase in H2O2 hydrogenation with addition of Au to heteropolyacid-only catalyst) at 2 °C (Table 1).| Catalyst | H2O2 Productivitya/mol kgcat−1 h−1 | H2O2 Hydrogenation b/mol kgcat−1 h−1 |
|---|---|---|
| a Rate of hydrogen peroxide production determined for reaction at 20 °C (all other conditions same as defined in Table 1). b Rate of hydrogenation of H2O2 calculated from amount of H2O2 hydrogenated at 20 °C (all other conditions same as defined in Table 1). | ||
| Cs2.8H0.2PW12O40 | 2 | 558 |
| Pd0.15Cs2.5H0.2PW12O40 | 43 | 670 |
| Au0.1Cs2.5H0.2PW12O40 | 35 | 282 |
| Pd0.1Au0.0333Cs2.5H0.2PW12O40 | 107 | 676 |
| Pd0.075Au0.05Cs2.5H0.2PW12O40 | 168 | 546 |
| 5% Pd/Cs2.8H0.2PW12O40 | 41 | 1105 |
| 5% Au/Cs2.8H0.2PW12O40 | 34 | 36 |
| 2.5% Au/2.5% Pd/Cs2.8H0.2PW12O40 | 30 | 1052 |
| 2.5% Au/2.5% Pd/C | 98 | 352 |
| 2.5% Au/2.5% Pd/C (2% HNO3) | 102 | 443 |
With the exception of the two Au–Pd-exchanged heteropolyacid and the Au-only heteropolyacid catalysts, all other catalysts showed lower H2O2 formation rates at ambient temperature (Table 2) compared to 2 °C (Table 1) and this is in agreement with the extent to which the H2O2 hydrogenation/decomposition rate over each catalyst increased at ambient temperature. Amongst all the Pd, Au and Au–Pd catalysts tested, Au–Pd/Cs2.8H0.2PW12O40 gives the best rate of H2O2 synthesis (198 mol kgcat−1 h−1) at 2 °C, but gave the lowest H2O2 productivity (30 mol kgcat−1 h−1) at ambient temperature and this correlates with its superior H2O2 hydrogenation/decomposition rate (1052 mol kgcat−1 h−1) at ambient temperature. Only 5% Pd/Cs2.8H0.2PW12O40 showed a slightly higher H2O2 hydrogenation rate than the Au–Pd/Cs2.8H0.2PW12O40. Interestingly, the use of ambient temperature enhanced H2O2 productivity over the Au–Pd-exchanged heteropolyacid and Au-only heteropolyacid (exchanged and supported) catalysts and the degree of enhancement was directly proportional to the Au content for the Au–Pd catalysts. Pd0.075Au0.05Cs2.5H0.2PW12O40 was the most effective catalyst, giving H2O2 productivity (168 mol kgcat−1 h−1) 4 times greater than the Pd-only exchanged heteropolyacid and significantly greater than Au–Pd/C(2% HNO3). In fact, the catalytic performance of this catalyst at ambient temperature was better than the performance of all the other catalysts (except 2.5% Au/2.5% Pd/Cs2.8H0.2PW12O40) at 2 °C. The catalyst also showed outstandingly higher TOF at ambient temperature (Fig. 2) than the supported heteropolyacid catalyst and the AuPd/C(2%HNO3) catalyst, which we have previously shown to be the most effective catalyst for H2O2 synthesis. Fig. 3 compares the change in H2O2 formation rate and wt% H2O2 formed at ambient temperature as a function of time for this Au–Pd-exchanged heteropolyacid catalyst and a conventional supported Au–Pd catalyst (Au–Pd/C). Both catalysts showed similar time online profiles; the H2O2 formed increased with time and attained a stable maximum before subsequently decreasing, while the rate was high initially and decreased continuously over time. However, the Au–Pd-exchanged heteropolyacid catalyst led to considerably higher H2O2 concentrations and rate of formation over the time-range investigated.
![A comparison of TOF for H2O2 synthesis at 20 °C using HPA-based and carbon-based Au, Pd and Au–Pd catalysts [data corrected to take account of contribution from the Au and Pd-free (heteropolyacid-only) material]. HPA – heteropolyacid, AW – acid washed.](/image/article/2012/GC/c1gc15863e/c1gc15863e-f2.gif) | ||
| Fig. 2 A comparison of TOF for H2O2 synthesis at 20 °C using HPA-based and carbon-based Au, Pd and Au–Pd catalysts [data corrected to take account of contribution from the Au and Pd-free (heteropolyacid-only) material]. HPA – heteropolyacid, AW – acid washed. | ||
 | ||
| Fig. 3 The effect of reaction time on the direct formation of H2O2 over Pd0.075Au0.05Cs2.5H0.2PW12O40 (solid lines) and Au–Pd/C (broken lines) using methanol and H2O as solvent at ambient temperature wt% H2O2 formed, H2O2 productivity. | ||


The observation that H2O2 productivity is only enhanced at room temperature over the Au-containing heteropolyacid catalysts and not the Pd-only heteropolyacid catalysts (show decrease in H2O2 productivity), suggest that Au plays a critical role in achieving the superior ambient temperature performance of the Au–Pd-exchanged heteropolyacid catalysts. The role of Au in promoting H2O2 formation over the Au–Pd-exchanged heteropolyacid and Au-only (exchanged and supported) catalysts at ambient temperature is mainly related to its ability to inhibit H2O2 hydrogenation/decomposition over these catalysts. This is supported by the fact that the Au-only (both supported and exchanged) heteropolyacid catalysts show a decrease in hydrogenation activity with increase in temperature from 2 °C to 20 °C and the degree of increase in hydrogenation activity with increasing temperature (2 °C as compared with 20 °C) over the Au–Pd-exchanged heteropolyacid catalysts is lower compared to the Pd-only exchanged heteropolyacid catalyst. Since the use of ambient temperature represents an important energy-saving option when compared with operation at sub-ambient temperatures, we consider the enhanced ambient temperature performance of the Au–Pd-exchanged heteropolyacid catalysts an interesting development for the direct synthesis of H2O2. In order to optimise hydrogen utilization, there is still a need to limit or completely switch-off the ambient temperature hydrogenation/decomposition of H2O2 over these catalysts and this is a subject of on-going research.
H2O2 synthesis and hydrogenation at ambient temperature with water as solvent using carbon and heteropolyacid-based Au, Pd and Au–Pd catalysts
Our standard reaction conditions (Table 1) make use of a mixture of methanol (66%) and H2O as solvent for the direct synthesis of H2O2. The presence of methanol improves the solubility of the reacting gases (H2 and O2) and stabilizes formed H2O2 against decomposition. However, most H2O2 applications require it diluted only in water. This means that if H2O2 is synthesised in H2O/methanol solvent, additional cost must be incurred to separate the methanol from the final product. Furthermore, H2O represents a non-toxic, non-flammable and cheaper solvent compared to alternative organic solvents and acids commonly used for the direct synthesis of H2O. We have therefore studied the effect of carrying out H2O2 synthesis and hydrogenation using H2O-only solvent at ambient temperature (all other reaction conditions remaining unchanged) and results are presented in Table 3. The H2O2 productivity over each catalyst (except the heteropolyacid-only catalyst) was considerably lower in H2O-only solvent (Table 3) compared to H2O/methanol solvent (Table 2). This can be attributed to the increase in H2O2 hydrogenation/decomposition observed upon using H2O-only solvent (see Tables 2 and 3) coupled with the lower solubility of H2 in H2O-only. The increase in H2O2 hydrogenation/decomposition using water as solvent confirms the role of methanol as a H2O2 stabiliser. The heteropolyacid-based catalysts prepared by ion exchange showed significantly higher H2O2 formation rates than all the Au-only, Pd-only and Au–Pd-supported heteropolyacid and carbon-based supported catalysts prepared by impregnation. Au–Pd-exchanged heteropolyacid catalysts (Pd0.1Au0.033Cs2.5H0.2PW12O40 and Pd0.075Au0.05Cs2.5H0.2PW12O40) again showed the best performance in H2O2 formation, with H2O2 productivity ca. 2 and 15 times higher than H2O2 productivity over the Pd-only exchanged heteropolyacid and all other Au–Pd-supported catalysts, respectively. The TOF in water at 20 °C (Table 4) for the best Au–Pd-exchanged heteropolyacid catalyst (Pd0.075Au0.05Cs2.5H0.2PW12O40) was over 140 times higher compared to the TOF for the conventionally supported Au–Pd/C(2% HNO3) catalyst previously shown to be the most active catalyst for H2O2 synthesis under our standard reaction conditions at 2 °C in water/methanol solvent. The heteropolyacid-only catalyst (Cs2.8H0.2PW12O40) showed a decrease in H2O2 hydrogenation using water as solvent and this was reflected in the significant increase (by an order of magnitude) in its H2O2 productivity. This suggests that in the absence of methanol (i.e. in H2O-only solvent), the heteropolyacid plays a crucial role in stabilising H2O2 from subsequent hydrogenation/decomposition. Indeed, the increase in H2O2 hydrogenation/decomposition in H2O-only solvent was higher for the carbon-based catalysts than for the heteropolyacid-based catalysts. Only the supported-Pd and Au–Pd heteropolyacid catalysts showed an increase in H2O2 hydrogenation comparable to the supported carbon catalysts (consistent with lower H2O2 productivities over these catalysts) and this can be explained by the fact that a higher number of the heteropolyacid sites in these catalysts are covered by Pd or Au–Pd sites.| Catalyst | H2O2 Productivitya/mol kgcat−1 h−1 | H2O2 Hydrogenation b/mol kgcat−1 h−1 |
|---|---|---|
| a Rate of hydrogen peroxide production determined for reaction at 20 °C using H2O (8.5 g) as solvent (all other conditions same as defined in Table 1). b Rate of hydrogenation of H2O2 calculated from amount of H2O2 hydrogenated at 20 °C using H2O (8.5 g) as solvent (all other conditions same as defined in Table 1). | ||
| Cs2.8H0.2PW12O40 | 22 | 282 |
| Pd0.15Cs2.5H0.2PW12O40 | 27 | 705 |
| Au0.1Cs2.5H0.2PW12O40 | 18 | 376 |
| Pd0.1Au0.0333Cs2.5H0.2PW12O40 | 58 | 793 |
| Pd0.075Au0.05Cs2.5H0.2PW12O40 | 61 | 776 |
| 5% Pd/Cs2.8H0.2PW12O40 | 4 | 1369 |
| 5% Au/Cs2.8H0.2PW12O40 | 16 | 217 |
| 2.5% Au/2.5% Pd/Cs2.8H0.2PW12O40 | 3 | 1310 |
| 2.5% Au/2.5% Pd/C | 4 | 799 |
| 2.5% Au/2.5% Pd/C (2% HNO3) | 4 | 746 |
| Catalyst | TOF/mol H2O2 molmetal−1 h−1 |
|---|---|
| a Data corrected to take account of contribution from the Au and Pd-free (heteropolyacid-only) material. Hydrogen peroxide production determined for reaction at 20 °C using H2O (8.5 g) as solvent (all other conditions same as defined in Table 1). | |
| Pd0.075Au0.05Cs2.5H0.2PW12O40 | 994 |
| 2.5% Au/2.5% Pd/Cs2.8H0.2PW12O40 | 8 |
| 2.5% Au/2.5% Pd/C (2% HNO3) | 11 |
Effect of CO2-free reaction gas (2% H2/air) on H2O2 synthesis and hydrogenation over carbon and heteropolyacid-based Au, Pd and Au–Pd catalysts
Our standard reaction gas for H2O2 synthesis (Table 1) comprise a mixture of 5% H2/CO2 and 25% O2/CO2 with H2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :O2 molar ratio of ca. 1:2. Although we have shown28 that dissolved CO2 gives the reaction solvent the acidity required to promote H2O2 formation, the use of CO2 diluent still represents an additional complexity. We have therefore investigated the direct synthesis of H2O2 using 2% H2/air as the reactant gas (Table 5). The H2O2 formation rate over each catalyst was considerably lower (when using 2% H2/air) compared to standard reaction conditions (Table 1). This is not surprising as the H2/O2 molar ratio (1/20) in 2% H2/air is markedly different from the 1/2 ratio employed using standard reaction conditions. This result is therefore consistent with our previous study,36 showing a decrease in H2O2 productivity with decreasing H2/O2 mol ratio. Au–Pd-exchanged and supported heteropolyacid catalysts were considerably more effective than the other catalysts investigated. The supported-heteropolyacid catalysts (2.5% Au/2.5% Pd/Cs2.8H0.2PW12O40 and 5% Pd/Cs2.8H0.2PW12O40) were the most active catalysts, showing slightly higher H2O2 productivity than the Au–Pd-exchanged heteropolyacid catalysts. We consider the Au–Pd-exchanged heteropolyacid catalysts to be the most efficient catalysts for H2O2 synthesis using 2% H2/air since they contain approximately an order of magnitude lower amount of metal, and, consequently, the Au–Pd-exchanged heteropolyacid catalysts were an order of magnitude more active than the Pd-only exchanged heteropolyacid catalyst. As already discussed (Table 2 and 3), these Au–Pd-exchanged heteropolyacid catalysts also showed superior performance for H2O2 synthesis at ambient temperature and with water as solvent when compared to the Pd-only exchanged heteropolyacid prepared using the same ion-exchange method. This indicates that the presence of Au in these heteropolyacid-based catalysts is key in achieving high H2O2 productivity under such green and cost-effective conditions (ambient temperature, water only solvent and the use of CO2-free reaction gas (2% H2/air)) preferred for industrial production of H2O2.
:O2 molar ratio of ca. 1:2. Although we have shown28 that dissolved CO2 gives the reaction solvent the acidity required to promote H2O2 formation, the use of CO2 diluent still represents an additional complexity. We have therefore investigated the direct synthesis of H2O2 using 2% H2/air as the reactant gas (Table 5). The H2O2 formation rate over each catalyst was considerably lower (when using 2% H2/air) compared to standard reaction conditions (Table 1). This is not surprising as the H2/O2 molar ratio (1/20) in 2% H2/air is markedly different from the 1/2 ratio employed using standard reaction conditions. This result is therefore consistent with our previous study,36 showing a decrease in H2O2 productivity with decreasing H2/O2 mol ratio. Au–Pd-exchanged and supported heteropolyacid catalysts were considerably more effective than the other catalysts investigated. The supported-heteropolyacid catalysts (2.5% Au/2.5% Pd/Cs2.8H0.2PW12O40 and 5% Pd/Cs2.8H0.2PW12O40) were the most active catalysts, showing slightly higher H2O2 productivity than the Au–Pd-exchanged heteropolyacid catalysts. We consider the Au–Pd-exchanged heteropolyacid catalysts to be the most efficient catalysts for H2O2 synthesis using 2% H2/air since they contain approximately an order of magnitude lower amount of metal, and, consequently, the Au–Pd-exchanged heteropolyacid catalysts were an order of magnitude more active than the Pd-only exchanged heteropolyacid catalyst. As already discussed (Table 2 and 3), these Au–Pd-exchanged heteropolyacid catalysts also showed superior performance for H2O2 synthesis at ambient temperature and with water as solvent when compared to the Pd-only exchanged heteropolyacid prepared using the same ion-exchange method. This indicates that the presence of Au in these heteropolyacid-based catalysts is key in achieving high H2O2 productivity under such green and cost-effective conditions (ambient temperature, water only solvent and the use of CO2-free reaction gas (2% H2/air)) preferred for industrial production of H2O2.
| Catalyst | H2O2 productivity/mol kg−1 h−1 | ||
|---|---|---|---|
| 2 °C, H2O + MeOHa | 20 °C, H2O + MeOHb | 20 °C, H2O-onlyc | |
| a Rate of hydrogen peroxide production determined for reaction using 2% H2/air under conditions that mimic standard reaction conditions (i.e. 4 MPa 2% H2/air, 8.5 g solvent (5.6 g MeOH + 2.9 g H2O), 0.01 g catalyst, 2 °C, 1200 rpm, 30 mins). b Rate of hydrogen peroxide production determined after reaction using 2% H2/air at 20 °C using a mixture MeOH and H2O as solvent (all other conditions unchanged) c Rate of hydrogen peroxide production determined after reaction using 2% H2/air at 20 °C using a H2O-only solvent (all other conditions unchanged). | |||
| Cs2.8H0.2PW12O40 | 0.7 | 8 | 6 |
| Pd0.15Cs2.5H0.2PW12O40 | 4 | 10 | 7 |
| Au0.1Cs2.5H0.2PW12O40 | 8 | 10 | 7 |
| Pd0.1Au0.033Cs2.5H0.2PW12O40 | 43 | 49 | 11 |
| Pd0.075Au0.05Cs2.5H0.2PW12O40 | 46 | 48 | 14 |
| 5% Pd/Cs2.8H0.2PW12O40 | 61 | 51 | 7 |
| 5% Au/Cs2.8H0.2PW12O40 | 6 | 14 | 6 |
| 2.5% Au/2.5% Pd/Cs2.5H0.2PW12O40 | 63 | 57 | 8 |
| 2.5% Au 2.5% Pd/C | 22 | 34 | 6 |
| 2.5% Au 2.5% Pd/C (HNO3) | 26 | 35 | 8 |
Using 2% H2/air as reactant led to similar effects for H2O2 synthesis as those observed using H2/CO2 and O2/CO2 with respect to the effect of solvent and reaction temperature. The data (Table 5) confirms that Au–Pd heteropolyacid-based catalysts are more efficient for the direct synthesis of H2O2 using green and cost-effective conditions (ambient temperature, H2O-only solvent) than Pd-only heteropolyacid and Au–Pd/C (untreated or acid-treated) catalysts
Effect of increase in Au–Pd loading and change in Cs content of Pd and Au–Pd-exchanged heteropolyacid catalysts
The Au, Pd and Au–Pd-exchanged heteropolyacid catalysts investigated so far only contain 0.49–0.61wt% Au, Pd or Au + Pd (Tables 1–3). We also investigated the effect of increasing the Pd and Au–Pd content of these catalysts through substitution of Cs. Pd and Au–Pd-exchanged heteropolyacid catalysts containing 2.11 and 2.35 wt% Pd and Au + Pd, respectively, were prepared and tested for H2O2 synthesis under standard conditions as well as using the challenging conditions (ambient temperature and water-only solvent). The results (Table 6) showed a considerable increase in H2O2 productivity (from 96 to 144 mol kg−1 h−1) with increasing Pd content for the Pd-only exchanged heteropolyacid when tested under standard conditions. However, as expected, catalysts with lower metal loading were more efficient in terms of TOFs. However, the higher loading catalyst at ambient temperature and in water-only solvent resulted in a decrease in activity. The Au–Pd-exchanged heteropolyacid catalyst showed a marked increase in activity under standard conditions (86–261 mol kg−1 h−1) and at ambient temperature (168–228 mol kg−1 h−1) with increasing Au–Pd content; the activity of the catalyst decreased from 61 to 40 mol kg−1 h−1 with increasing Au–Pd content in water-only solvent. The observed trends in activity appear to be related to the acidity of these catalysts and this is discussed in a subsequent section. Although the higher loading Au–Pd-exchanged heteropolyacid catalyst shows considerably higher H2O2 synthesis activity (228 mol kgcat−1 h−1) at ambient temperature in water/methanol solvent, we do not observe the increase in activity noted for the lower loading catalysts as the temperature is increased from 2 °C to ambient temperature as there is a decrease from 261 to 221 mol kgcat−1 h−1. However, the extent of the decrease in activity is considerably lower when compared to the higher loading Pd-only exchanged heteropolyacid catalyst. The absence of activity enhancement with an increase in temperature for the higher loaded Au–Pd-exchanged heteropolyacid catalyst can be related to these high activity catalysts being more active for hydrogenation of H2O2.| Catalyst | Metal wt (%) | H2O2 Productivity/mol kgcat−1 h−1 | ||
|---|---|---|---|---|
| 2 °C, water + MeOHa | 20 °C, water + MeOHb | 20 °C, water onlyc | ||
| a , b Rate of hydrogen peroxide production determined for reaction at 2 °Ca or 20 °Cb using standard reaction conditions: 5% H2/CO2 (2.9 MPa) and 25% O2/CO2 (1.1 MPa), 8.5 g solvent (5.6 g MeOH + 2.9 g H2O), 0.01 g catalyst, 1200 rpm, 30 min). c Rate of hydrogen peroxide production determined after reaction at 20 °C using water-only solvent. Reaction conditions: 5% H2/CO2 (2.9 MPa) and 25% O2/CO2 (1.1 MPa), 8.5 g H2O, 0.01 g catalyst, 1200 rpm, 30 min). | ||||
| Pd0.15Cs2.5H0.2PW12O40 | 0.49 | 96 | 43 | 27 |
| Pd0.65Cs1.5H0.2PW12O40 | 2.11 | 144 | 37 | 13 |
| Pd0.075Au0.05Cs2.5H0.2PW12O40 | 0.53 | 86 | 168 | 61 |
| Pd0.325Au0.217Cs1.5H0.2PW12O40 | 2.35 | 261 | 228 | 40 |
Catalyst characterisation
IR spectra recorded for the heteropolyacid catalysts (Fig. 4a and 4b) showed the characteristic features of phosphotungstic acids comprising the Keggin structure.37 The Keggin structures characteristic vibrational bands were observed at 1079 cm−1 [νas(P–O) vibration], 975 cm−1 [terminal νas(W![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) vibration], 887 and 795 cm−1 [assigned to two different νas(W–O–W) vibrations]. The weak bands at 2400 cm−1 relate to CO2 from the air. Some spectra also show bands centred at ca. 3400 cm−1, which can be assigned to ν(O–H). The ν(O–H) vibrational bands may be due to water adsorbed on the sample. Some samples also show a band at 1710 cm−1 characteristic of the presence of protonated water clusters, such as H5O2+. Since the spectra were recorded in open air without further drying of samples prior to the recording of spectra, these (O–H) vibrations cannot be exclusively assigned to the heteropolyacid structure. In all samples after Cs+ substitution the P–O stretch remains constant, indicating that the substitution of Cs+ for H+ does not cause strain on the P–O4 tetrahedron at the centre of the structure. In accordance with the literature37 a shift in νas(WO) from 975 to 982 cm−1 on incorporation of Cs+ (spectra B–F) indicates a strengthening of WO bonds. This indicates that less hydrogen bonding is present, caused by the removal of H+ from the Keggin structure. The IR spectra recorded do not give rise to any new features on incorporation of Au and Pd by impregnation or ion exchange. However, since only small amounts of Au and Pd are incorporated by ion exchange any new signals may be below the limit of detection. The IR spectra of the impregnated catalysts (spectra C and D) show that on impregnation of 5 wt% Pd and 2.5% Pd 2.5% Au followed by calcination, the Keggin structure has been maintained. IR spectroscopy also shows that the Keggin structure was maintained when using ion exchange preparation methods.
O) vibration], 887 and 795 cm−1 [assigned to two different νas(W–O–W) vibrations]. The weak bands at 2400 cm−1 relate to CO2 from the air. Some spectra also show bands centred at ca. 3400 cm−1, which can be assigned to ν(O–H). The ν(O–H) vibrational bands may be due to water adsorbed on the sample. Some samples also show a band at 1710 cm−1 characteristic of the presence of protonated water clusters, such as H5O2+. Since the spectra were recorded in open air without further drying of samples prior to the recording of spectra, these (O–H) vibrations cannot be exclusively assigned to the heteropolyacid structure. In all samples after Cs+ substitution the P–O stretch remains constant, indicating that the substitution of Cs+ for H+ does not cause strain on the P–O4 tetrahedron at the centre of the structure. In accordance with the literature37 a shift in νas(WO) from 975 to 982 cm−1 on incorporation of Cs+ (spectra B–F) indicates a strengthening of WO bonds. This indicates that less hydrogen bonding is present, caused by the removal of H+ from the Keggin structure. The IR spectra recorded do not give rise to any new features on incorporation of Au and Pd by impregnation or ion exchange. However, since only small amounts of Au and Pd are incorporated by ion exchange any new signals may be below the limit of detection. The IR spectra of the impregnated catalysts (spectra C and D) show that on impregnation of 5 wt% Pd and 2.5% Pd 2.5% Au followed by calcination, the Keggin structure has been maintained. IR spectroscopy also shows that the Keggin structure was maintained when using ion exchange preparation methods.
 | ||
| Fig. 4 (a) IR spectra between 4000–500 cm−1 of A – H3PW12O40·6H2O, B – Cs2.8H0.2PW12O40, C – 5% Pd/Cs2.8H0.2PW12O40, D – 2.5%Au/2.5%Pd/Cs2.8H0.2PW12O40, E – Pd0.15Cs2.5H0.2PW12O40 and F – Pd0.075Au0.05Cs2.5H0.2PW12O40. (b) IR spectra between 1500–500 cm−1 of A – H3PW12O40·6H2O, B – Cs2.8H0.2PW12O40, C – 5% Pd/Cs2.8H0.2PW12O40, D – 2.5% Au/2.5% Pd/Cs2.8H0.2PW12O40, E – Pd0.15Cs2.5H0.2PW12O40 and F – Pd0.075Au0.05Cs2.5H0.2PW12O40 showing the shift in WO peak at 975 cm−1. | ||
Raman spectra (Fig. 5) recorded for Au, Pd and Au–Pd heteropolyacid catalysts show Raman bands related to atomic vibrations characteristic of peroxocomplex fragments38 in the range of Raman shifts of 0–1200 cm−1. Raman bands at 996 and 1006 cm−1 are associated with v(WO) bond vibrations while a single band at 915 cm−1 corresponds to the v(O–O) vibration set. A broader band at 550 cm−1 has been assigned to two possible features; specifically asymmetric and/or symmetry vibrational modes of the W–(O2) bond. In accordance with the literature,38 bands located at lower frequencies (236, 216, 160, 154 and 109) arise from the deformation of complex fragments in the heteropolyacid framework. Additional Raman bands centred at 318 and 342 cm−1 have been identified for the 5% Au/Cs2.5H0.2PW12O40catalyst prepared by impregnation, and these probably relate to vibrations from a Au-species. It is possible from closer inspection of the spectra to surmise that the decrease in resolution for bands centred at 540 and 915 cm−1 are due to interaction of Au atoms with oxygen defects and termination points. In summary, the spectra for the heteropolyacid series shows that the key characteristic heteropolyacid bands are retained for all catalysts irrespective of the incorporation of Au, Pd or Au–Pd by ion-exchange or impregnation. Only the Au-heteropolyacid catalyst prepared by impregnation (containing ca. 8 times more Au than catalysts prepared by ion-exchange) shows additional features that might be related to the presence of Au. Pd-based heteropolyacid prepared by impregnation or ion-exchange showed no additional Raman bands.
 | ||
| Fig. 5 Raman spectra recorded for a series of heteropolyacid based catalysts prepared by ion-exchange and impregnation methods. Wt% loadings of Au and Pd range from 0.01 to 5% for catalysts: A – Cs2.8H0.2PW12O40, B – Pd0.65Cs1.5H0.2PW12O40, C – Pd0.15Cs2.5H0.2PW12O40, D – Pd0.10Au0.03Cs2.5H0.2PW12O40, E – Pd0.075Au0.05Cs2.5H0.2PW12O40 and F – 5% PdCs2.5H0.2PW12O405% Au/Cs2.5H0.2PW12O40. | ||
All samples (whether with Pd and Au added by either impregnation or ion exchange methods) show similar XRD spectra (Fig. 6a–c). The detected reflections are consistent with the cubic structure of the Cs3PW12O40 salt (ICDD number 00-051-1857) as the catalysts prepared have similar Cs content. The XRD spectra of the Au and Pd-supported Cs2.8H0.2PW12O40 catalysts do not give rise to any changes to the reflections associated with the Cs salt. The 5% Pd supported catalyst does not show any new reflections that could be assigned to either metallic Pd or PdO, this could be attributed to the very small crystallite size of these Pd or PdO species. The XRD spectra of 5% Au supported catalyst shows reflections at 2θ values (38, 44, 64 and 77°) characteristic of metallic Au. Both monometallic and bimetallic Au and Pd ion-exchanged catalysts showed no new features when compared to Cs2.8H0.2PW12O40. This indicates that the structure of the Cs-substituted salt remained intact after ion exchange. The XRD spectra of the catalyst with lower Cs content, Pd0.075Au0.05Cs1.5H0.2PW12O40, showed peaks associated with Cs2.8H0.2PW12O40 along with some new weak features at 2θ = 15 and 45°assigned to the presence of H3PW12O40 (see Fig. 6c). The structure of this lower Cs-containing catalyst (Pd0.075Au0.05Cs1.5H0.2PW12O40) lies in between the structure of Cs3PW12O40 and H3PW12O40.
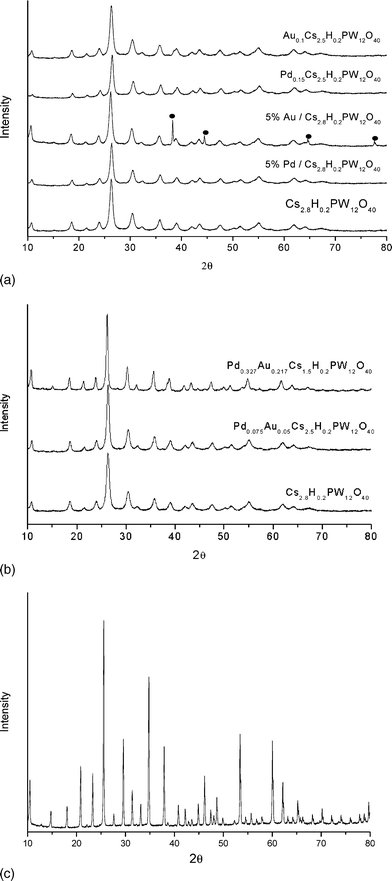 | ||
| Fig. 6 Powder X-ray diffraction paterns for (a) Au-only and Pd-only HPA-based catalysts, (b) Au–Pd HPA-based catalysts, (c) H3PW12O40. | ||
NH3-TPD profiles (Fig. 7) recorded for Pd, Au and Au–Pd heteropolyacid catalysts prepared by ion-exchange confirmed that all catalysts have acidic character with the amount and strength of acid sites depending on both the Cs content and the type(s) of other metals (Au, Pd or Au + Pd) incorporated into the structure (see Table 7). Catalysts containing Cs2.5 all show a small ammonia desorption at around 800 °C compared to catalysts containing less cesium, Cs1.5, which show a large desorption at 570 °C, which is around four times larger. This indicates that lower Cs content leads to more acid sites, which are weaker in strength than Cs2.5 containing catalysts. On incorporation of metals into the heteropolyacid structure the acidity is modified, it appears that for Cs2.5 catalysts incorporation of one metal slightly reduces the acidity when compared to the support material, Cs2.5H0.5PW12O40. However, on incorporation of both Au and Pd more ammonia is desorbed from the catalyst, showing a slight increase in acidity. This is also true for the Cs1.5 containing catalysts, with relative NH3 uptakes increasing from 3.57 to 4.73 on incorporation of both Au and Pd when compared to a similar metal loading of Pd only. As noted earlier, the observed trends in activity appear to be related to the acidity of these catalysts. The higher loading Au–Pd catalysts have a higher number of acid sites that contribute to their higher activity under standard conditions and when using ambient temperature in water/methanol solvent. At ambient temperature in water-only solvent, the activity of the higher loading catalyst decreases because, although there are a larger number of acid sites, the catalysts comprise weaker acid sites compared to the lower loaded catalysts. In the absence of methanol (i.e. in water-only solvent), a strong acidic catalyst is required to stabilise the formed H2O2 from subsequent decomposition/hydrogenation.
 | ||
| Fig. 7 TPD profiles of ion-exchanged HPA-based catalysts. | ||
XPS data (Table 8) shows the presence of surface Pd and/or Au species for Pd-only, Au-only and Au–Pd heteropolyacid catalysts prepared by impregnation. Where possible we also give the data for the oxidation states of the Au and Pd, although in some of the catalysts the amount of these metals present is too little to be able to unambiguously determine the oxidation state. The Au–Pd/heteropolyacid catalyst shows a Pd/Au ratio of ca. 5.7, which suggests the formation of Au–Pd nanoparticles with a core–shell structure (Au-rich core and Pd-rich surface). With the exception of the Pd-only ion-exchanged catalyst (which showed surface Pd species), XPS data for the Au-only and Au–Pd heteropolyacid catalysts prepared by ion-exchange did not show any surface Au and/or Pd species. While the absence of these surface species might be related to the relatively low loading (0.5–0.6 wt%) of Au or Au + Pd in these catalysts, the fact that we observe surface Pd species for the Pd-only ion-exchanged heteropolyacid catalyst (which also has relatively very low Pd loading, 0.49 wt%) might suggest that most of the Au and/or Pd species present in the Au-only and Au–Pd ion-exchanged heteropolyacid catalysts are not on the surface but incorporated within the heteropolyacid porous structure. It is possible that the presence of Au together with Pd leads to the formation of Au–Pd nanoparticles, which are much smaller (than the Pd-only particles) and can therefore be easily incorporated into the porous framework. It is clear, however, that the addition of Au to these catalysts has a marked effect on the catalysis. As the gold is not on the surface it may be that the effect we observe is similar to that we have observed for supported alloy nanoparticles with Pd-rich shells and Au-rich cores for oxide-supported catalysts.39 However, attempts to characterise the heteropolyacid catalysts used in this study with electron microscopy have not proved feasible due to the extreme beam-sensitivity of these materials.
| Surface analysis | ||||||
|---|---|---|---|---|---|---|
| Sample | Au/Pd oxidation state | Stoichiometry | ||||
| Pd | Au | Cs | P | W | ||
| a nd = not detected by XPS, the metal content was too low for this method. | ||||||
| Pd0.15Cs2.5H0.2PW12O40 | Pd2+(100%) | 0.20 | 0 | 2.02 | 0.98 | 12.0 |
| Pd0.1Au0.0333Cs2.5H0.2PW12O40 | nda | 2.00 | 1.49 | 12.0 | ||
| Pd0.075Au0.05Cs2.5H0.2PW12O40 | nd | 2.12 | 1.17 | 12.0 | ||
| Cs2.8H0.2PW12O40 | — | — | — | 2.50 | 1.08 | 12.0 |
| 2.5% Au/2.5% Pd/Cs2.8H0.2PW12O40 | Pd2+(43%) Pd0 (57%) Au0(100%) | 0.86 | 0.15 | 5.21 | 2.03 | 12.0 |
| 5% Pd/Cs2.8H0.2PW12O40 | Pd2+(85%) Pd0 (15%) | 1.94 | 1.09 | 12.0 | ||
| 5% Au/Cs2.8H0.2PW12O40 | Au0 (100%) | 2.07 | 2.25 | 12.0 | ||
| Au0.1Cs2.5H0.2PW12O40 | nd | 1.64 | 1.80 | 12.0 | ||
Conclusions
Pd-only, Au-only and Au–Pd-exchanged and supported heteropolyacid catalysts have been investigated for the direct synthesis of H2O2 from H2 and O2. The heteropolyacid-based Au–Pd catalysts are more effective for H2O2 formation than corresponding Pd-only catalysts prepared using the same impregnation or ion-exchanged method. The supported Au–Pd heteropolyacid catalyst (2.5% Au/2.5% Pd/Cs2.8H0.2PW12O40) was the most active catalyst under standard reaction conditions. This catalyst showed significantly higher H2O2 productivity than the most effective catalyst (2.5% Au/2.5% Pd/C (2% HNO3) we have previously reported for the direct synthesis of H2O2 using standard reaction conditions. The catalyst also showed the best H2O2 synthesis activity when using 2% H2/air for direct H2O2 synthesis. The Au–Pd-exchanged heteropolyacid catalysts (Pd0.1Au0.0333Cs2.5H0.2PW12O40 and Pd0.1Au0.0333Cs2.5H0.2PW12O40) were slightly less active for H2O2 synthesis using 2% H2/air than the Au–Pd-supported heteropolyacid catalyst but showed superior activity compared to the Pd-only exchanged heteropolyacid (Pd0.15Cs2.5H0.2PW12O40) and supported Au–Pd/C (untreated and acid-treated) catalysts. Although the Au–Pd-exchanged heteropolyacid catalysts showed comparable activity to the Pd-only heteropolyacid catalyst and were less active than Au–Pd/C (2% HNO3) and Au–Pd/Cs2.8H0.2PW12O40 under standard reaction conditions, these catalysts proved to be most effective for H2O2 synthesis at ambient temperature and when using H2O-only solvent. Since the Au–Pd-exchanged heteropolyacid catalysts only contain ca. 3% of the total Au and Pd metal content present in the more conventional supported Au–Pd heteropolyacid and carbon catalysts, we consider that these catalysts could be the basis of more efficient catalysts for the direct H2O2 synthesis at ambient temperature and using air as compared with oxygen. It should be noted that the presence of Au in these heteropolyacid-type catalysts is crucial in achieving superior catalytic performance under these reaction conditions. Based on environmental and process economics considerations, the use of ambient temperature, water-only solvent and 2% H2/air for industrial production of H2O2 will be preferred to low temperature (2 °C), methanol-containing solvent and 5% H2/CO2 + 25% O2/CO2 respectively. However, it should be noted that the over-riding consideration for the commercialisation of catalysts for the direct synthesis of hydrogen peroxide is the selectivity of the process and this is a factor that will require further improvement with these new heteropolyacid-supported catalysts. We therefore consider the improved performance of the Au–Pd-exchanged heteropolyacid catalysts under these preferred conditions an important advance in the development of catalysts for the direct synthesis of H2O2.Acknowledgements
We thank the EPSRC for financial support.References
- C. W. Corti, R. J. Holliday and D. T. Thompson, Appl. Catal., A, 2005, 291, 253 CrossRef CAS.
- http://news.bbc.co.uk/1/hi/england/london/4197500.stm .
- H. Henkel and W. Weber, US Pat., 1108752, 1914.
- G. A. Cook, US Pat., 2368640, 1945.
- A. I. Dalton, E. J. Greskovich and R. W. Skinner, US Pat., 4336238, 1982.
- L. W. Gosser, US Pat., 4889705, 1989.
- H. Nagashima, Y. Ishiuchi and Y. Hiramatsu, US Pat., 5236692, 1993.
- K. T. Chuang and B. Zhou, US Pat., 5925588, 1999.
- D. P. Dissanayake and J. H. Lunsford, J. Catal., 2002, 206, 173 CrossRef CAS.
- J. H. Lunsford, J. Catal., 2003, 216, 455 CrossRef CAS.
- S. Chinta and J. H. Lunsford, J. Catal., 2004, 225, 249 CrossRef CAS.
- Y. Han and J. H. Lunsford, J. Catal., 2005, 230, 313 CrossRef CAS.
- V. R. Choudhary and C. Samanta, J. Catal., 2006, 238, 28 CrossRef CAS.
- Q. Liu and J. H. Lunsford, J. Catal., 2006, 239, 237 CrossRef CAS.
- V. R. Choudhary and P. Jena, J. Catal., 2007, 246, 434 CrossRef CAS.
- C. Samanta and V. R. Choudhary, Catal. Commun., 2007, 8, 73 CrossRef CAS.
- C. Samanta and V. R. Choudhary, Appl. Catal., A, 2007, 330, 23 CrossRef CAS.
- V. R. Choudhary, C. Samanta and T. V. Choudhary, Catal. Commun., 2007, 8, 1310 CrossRef CAS.
- V. R. Choudhary, C. Samanta and P. Jena, Appl. Catal., A, 2007, 332, 70 CrossRef CAS.
- C. Samanta and V. R. Choudhary, Chem. Eng. J., 2008, 136, 126 CrossRef CAS.
- P. Landon, P. J. Collier, A. J. Papworth, C. J. Kiely and G. J. Hutchings, Chem. Commun., 2002, 2058 RSC.
- P. Landon, P. J. Collier, A. F. Carley, D. Chadwick, A. J. Papworth, A. Burrows, C. J. Kiely and G. J. Hutchings, Phys. Chem. Chem. Phys., 2003, 5, 1917 RSC.
- J. K. Edwards, B. Solsona, P. Landon, A. F. Carley, A. Herzing, M. Watanabe, C. J. Kiely and G. J. Hutchings, J. Mater. Chem., 2005, 15, 4595 RSC.
- J. K. Edwards, B. Solsona, P. Landon, A. F. Carley, A. Herzing, M. Watanabe, C. J. Kiely and G. J. Hutchings, J. Catal., 2005, 236, 69 CrossRef CAS.
- A. Lopez-Sanchez, N. Dimitratos, P. Miedziak, E. Ntainjua, J. K. Edwards, D. Morgan, A. Carley, R. Tiruvalam, C. J. Kieley and G. J. Hutchings, Phys. Chem. Chem. Phys., 2008, 10, 1921 RSC.
- J. K. Edwards, A. F. Carley, A. Herzing, C. J. Kiely and G. J. Hutchings, Faraday Discuss., 2007, 138, 225 RSC.
- G. Li, J. Edwards, A. F. Carley and G. J. Hutchings, Catal. Today, 2007, 122, 361 CrossRef CAS.
- J. K. Edwards, A. Thomas, A. F. Carley, A. Herzing, C. J. Kiely and G. J. Hutchings, Green Chem., 2008, 10, 388 RSC.
- E. Ntainjua, N. J. K. Edwards, A. F. Carley, J. A. Lopez-Sanchez, J. A. Moulijn, A. A. Herzing, C. J. Kiely and G. J. Hutchings, Green Chem., 2008, 10, 1162 RSC.
- G. J. Hutchings, Chem. Commun., 2008, 1148 Search PubMed.
- E. Ntainjua, N. M. Piccinini, J. C. Pritchard, J. K. Edwards, A. F. Carley, J. A. Moulijn and G. J. Hutchings, ChemSusChem, 2009, 2, 575 CrossRef.
- E. Ntainjua, N. M. Piccinini, J. C. Pritchard, J. K. Edwards, A. F. Carley, J. A. Moulijn, C. J. Kiely and G. J. Hutchings, ChemCatChem., 2009, 1, 479–484 CrossRef.
- J. K. Edwards, B. Solsona, E. Ntainjua, N. A. F. Carley, A. A. Herzing, C. J. Kiely and G. J. Hutchings, Science, 2009, 323, 1037 CrossRef CAS.
- M. Sun, J. Zhang, Q. Zhang, Y. Wang and H. Wan, Chem. Commun., 2009, 5174 RSC.
- S. Park, S. H. Lee, S. H. Song, D. R. Park, S. H. Baeck, T. J. Kim, Y-M. Chung, S-H. Oh and I. K. Song, Catal. Commun., 2009, 10, 391 CrossRef CAS.
- M. Piccinini, E. Ntainjua, J. K. Edwards, A. F. Carley, J. A. Moulijn and G. J. Hutchings, Phys. Chem. Chem. Phys., 2010, 12, 2488 RSC.
- N. Essayem, A. Holmqvist, P. Y. Gayraud, J. C. Vedrine and Y. Ben Taarit, J. Catal., 2001, 197, 273 CrossRef CAS.
- Z. P. Pai, D. I. Kochubey, P. V. Berdnikova, V. V. Kanazhevskiy, I. Y. Prikhod'ko and Y. A. Chesalov, J. Mol. Catal. A: Chem., 2010, 332, 122 CrossRef CAS.
- D. I Enache, J. K. Edwards, P. Landon, B. Solsona-Espriu, A. F. Carley, A. A. Herzing, M. Watanabe, C. J. Kiely, D. W. Knight and G. J. Hutchings, Science, 2006, 311, 362 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |