Selective catalytic etherification of glycerol formal and solketal with dialkyl carbonates and K2CO3†
Maurizio
Selva
*,
Vanni
Benedet
and
Massimo
Fabris
Dipartimento di Scienze Molecolari e Nanosistemi dell'Università Ca' Foscari Venezia, Calle Larga S. Marta, 2137-30123, Venezia, Italy. E-mail: selva@unive.it; Fax: +39 041 234 8958; Tel: +39 041 234 8687
First published on 16th November 2011
Abstract
At T ≥ 200 °C, in the presence of K2CO3 as a catalyst, an original etherification procedure of non-toxic acetals such as glycerol formal (GlyF) and solketal has been investigated by using dialkyl carbonates as safe alkylating agents. The effects of parameters including the temperature, the reaction time, and the loading of both the catalyst and the dialkyl carbonate have been detailed for the model case of dimethyl carbonate (DMC). Both GlyF and solketal were efficiently alkylated by DMC to produce the corresponding O-methyl ethers with selectivity up to 99% and excellent yields (86–99%, by GC). The high selectivity could be accounted for by a mechanistic study involving a combined sequence of methylation, carboxymethylation, decarboxylation and hydrolysis processes. The O-methylation of GlyF and solketal could be successfully scaled up for multigram synthesis even operating with a moderate excess (5 molar equiv.) of DMC and in the absence of additional solvent. Notwithstanding the advantageous reduction of the process mass index, scale up experiments provided evidence that prolonged reaction times may induce the decomposition of DMC mainly by the loss of CO2. The K2CO3-catalyzed etherification of solketal with other carbonates such as dibenzyl and diethyl carbonate (DBnC and DEC, respectively), proceeded with the same good selectivity observed for DMC. However, at 220 °C, the solketal conversion did not exceed 81% since both DBnC and DEC were extensively consumed in competitive decarboxylation and hydrolysis reactions.
Introduction
Glycerol is a non-toxic biodegradable compound whose relevance as a chemical is recognized worldwide in a number of applications for the pharma, polymer, and food sectors.1 Not by chance, in 2004, the National Laboratories of the US Department of Energy, have classified glycerol among the twelve most attractive platform chemicals that can be produced from biomass via biological or chemical conversions.2 However, due to the exponential increase of biodiesel production and demand, the global market is experiencing a massive surplus of glycerol: in the EU for example, some additional 1.0 × 106 tonnes of such a compound have been available in 2010 (almost tripled since 2005).3These considerations have fuelled a growing interest of both academic and industrial communities for intensive research programs aimed at the conversion of glycerol and its derivatives into energy and chemicals. In this context, glycerol acetals, particularly light compounds such as glycerol formal (GlyF) and solketal (1a–b and 1c, respectively) recently caught our attention (Scheme 1). Compounds 1 are non-toxic substances synthesized by acid-catalysed condensations of glycerol with formaldehyde and acetone.4 GlyF is commercially available as a mixture of two 5- and 6-membered ring isomers (1a and 1b in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 molar ratio, respectively), while solketal is obtained exclusively as a five-membered ring product.5
:3 molar ratio, respectively), while solketal is obtained exclusively as a five-membered ring product.5
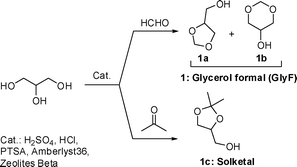 | ||
| Scheme 1 Light glycerol acetals. | ||
As such, both acetals are mainly used in the field of solvents for injectable pharmaceutical preparations, paints and water-based inks,4c,6 and for the preparation of additives for biodiesel formulations.3a,5d,7 GlyF and solketal however, are also valuable glycerol synthons whose reactivity involves almost exclusively the conversion of their free hydroxyl functionality by conventional esterification and etherification agents (carboxylic acids and alkyl halides, respectively).8,9 This aspect prompted us to investigate whether eco-friendly protocols could be devised for such processes, especially for the synthesis of O-alkyl derivatives of both 1a–b and 1c. To this purpose, dialkyl carbonates (DAlCs) appeared attractive compounds since they offer genuine green advantages over classical alkylating reagents:10i) they are non-toxic compounds; ii) their alkylation mediated reactions are always catalytic processes, with no by-products except for alcohols recyclable to the synthesis of DAlCs, and CO2 which does not involve disposal problems; iii) very often, (light) organic carbonates can be used not only as reactants, but also as reaction solvents. Moreover, their alkylation reactivity can be tuned for a variety of nucleophiles. This behaviour has been extensively investigated by our research group in the past fifteen years.11 Consider, for example, the model case of base-catalysed methylations mediated by dimethyl carbonate (Scheme 2).
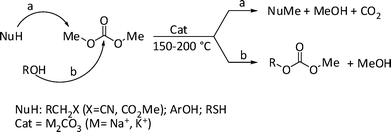 | ||
| Scheme 2 | ||
At 150–200 °C, in the presence of solid alkaline carbonates as catalysts, the reaction of CH2-active compounds, phenols, and thiols (NuH) with DMC proceeds with an exceptionally high selectivity (>99%) towards the corresponding methyl derivatives (NuMe, path a).
Under these conditions, a different scenario is offered by alcohols: plausibly due to their hard nucleophilic character, a number of primary and secondary alcohols including benzyl substrates, react with DMC to produce predominantly the corresponding transesterification products (path b).11h,12 Notwithstanding the alcohol-like reactivity expected for glycerol acetals, this paper demonstrates that reaction conditions can be found for the selective etherification of the hydroxyl group of both GlyF and solketal with DMC. In particular, this unprecedented reaction takes place at 200–220 °C, in the presence of potassium carbonate as a catalyst (Scheme 3).
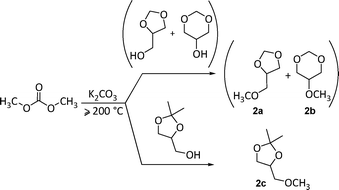 | ||
| Scheme 3 | ||
An in-depth investigation has been carried out to analyze the course of the overall transformation. Beyond the O-methylation process which affords methyl ethers 2 in yields up to 90%, results provide evidence that also the transesterification of acetals 1a–b and 1c with DMC takes place, producing the corresponding carboxymethyl derivatives (Scheme 2, path b). These compounds however, disappear as the reaction proceeds: both the reversible nature of the transesterification and the occurrence of competitive processes of decarboxylation and hydrolysis of carboxymethyl products account for this result. Accordingly, a mechanistic hypothesis has been formulated.
The same mechanism plausibly holds true for higher homologues of DMC: both diethyl- and dibenzyl- carbonate react with solketal to yield the related carboxyalkyl and O-alkyl derivatives, the latter compounds being the final reaction products. Although an excellent alkylation selectivity (up to 100%) can be achieved, these transformations appear somewhat limited by the consumption of the starting dialkyl carbonatesvia concurrent decarboxylation and hydrolysis processes.
Overall, the catalytic O-alkylation of renewable acetals of glycerol with safe dialkyl carbonates, particularly DMC, not only represents a genuinely green procedure, but it unlocks a potential for preparative purposes: this study demonstrates that the reaction can be scaled up for multigram synthesis without any loss of selectivity.
Results
Since previous studies on base catalysed reactions of GlyF and solketal with dialkyl carbonates were not available, a full set of experiments was necessary to examine the feasibility of the process. Our experience on the alkylation reactivity of DMC steered us on the choice of reaction conditions, particularly on the range of major parameters such as catalyst amount, temperature, time, and reactants molar ratios.The reaction of glycerol formal with DMC in the presence of K2CO3
Initial experiments were carried out in a stainless steel autoclave (200 mL) charged with a mixture of commercial glycerol formal (isomers 1a–1b in a 2:3 ratio; 0.80 g; 7.7 mmol), dimethyl carbonate (DMC, 19 mL, molar ratio W = DMC:GlyF = 30) and K2CO3 (molar ratio Q = K2CO3:GlyF = 1.2). DMC was used in a large excess, acting simultaneously as a reagent and a solvent. The autoclave was purged with N2, electrically heated at 200 °C, and kept under magnetic stirring. Reactions were followed for different time intervals of 6, 10, 20 and 24 h, respectively. At the end of each experiment, the reactor was cooled to rt and vented, and the reaction mixture was analyzed by GC-MS. In all cases, major products (≥95%) derived from O-methylation and carboxymethylation reactions of GlyF (compounds 2a–2b and 3a–3b, respectively; Scheme 4).
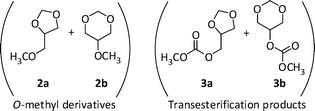 | ||
| Scheme 4 Major products of the K2CO3-catalysed reaction of glycerol formal with DMC. | ||
Products 2 and 3 were isolated and fully characterized by GC-MS, 1H and 13C NMR (further details are in the experimental section). The structure of isomers 3a–3b was also confirmed by comparison to authentic samples. Other unidentified by-products (≤5% in total, by GC), were also detected. The results are shown in Fig. 1a–b where the amount (%, by GC) of both reagents and products is plotted vs. the reaction time.13
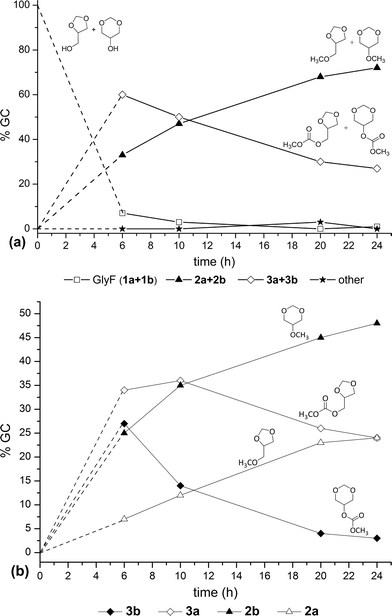 | ||
| Fig. 1 The reaction of GlyF with DMC carried out at 200 °C: (a) total amounts of each couple of isomers (1a+1b, 2a+2b, 3a+3b, respectively). “Other” were unidentified by-products; (b) amounts of each single isomer of methyl (2a–2b) and carboxymethyl (3a–3b) products. | ||
Four main aspects emerged from Fig. 1a: i) after 10 h, the reaction conversion was substantially quantitative (97%); ii) the amount of O-methylation products (2a+2b) increased progressively with time, with a methylation selectivity up to 73% after 24 h; iii) the amount of carboxymethylation products (3a+3b) went up to a maximum in the first 6 h, and then, it dropped to 27% as the reaction proceeded further; iv) by-products were negligible. Fig. 1b showed that six-membered ring products (O-methyl and carboxymethyl derivatives 2b and 3b) formed and reacted, respectively, faster than their five-membered ring analogues (O-methyl and carboxymethyl derivatives 2a and 3a). After 24 h, the amount of 2b was doubled than 2a (24 and 48%, respectively); while, compound 3b and 3a were present in 24 and 3% amounts, respectively.
Once the last experiment (24 h reaction) of Fig. 1 was concluded, K2CO3 was filtered and dried under vacuum overnight (8 mbar, 100 °C). The solid was finely powdered and reused to catalyze the reaction of GlyF and DMC under the conditions of Fig. 1 (24 h). The recycle of K2CO3 was repeated once more. GC-MS analyses of experiments with both fresh and recycled catalyst showed conversions and product distributions that did not differ by more than 5% from each other. This confirmed that K2CO3 was a recyclable catalyst. The result matched the behaviour already reported for DMC mediated carboxylations and methylations of different nucleophiles.14
Two additional experiments were carried out in the absence of K2CO3. A mixture of GlyF and DMC was set to react for 10 and 24 h under the conditions of Fig. 1. The conversions of GlyF were 60 and 96%, respectively, but only a transesterification reaction took place yielding carboxymethylated compounds (3a+3b) as exclusive products. This in line with literature results for the thermal (non-catalytic) transesterifications of esters and organic carbonates which were reported at T > 170 °C.15
Alkali metal exchanged faujasites could also act as efficient catalysts for O-alkylation of alcohols with dialkyl carbonates.11 However, attempts to use faujasites in the reaction of GlyF and solketal with DMC, were not successful. For example, at 200 °C, the conversion of GlyF was complete after only 3 h on NaX (weight ratio zeolite:GlyF = 1), but the formation of O-methyl derivatives 2a+2b (72%, by GC) was accompanied by the aperture of the cyclic structures of both GlyF and products 2, which yielded an extensive amount of by-products (28%). This competitive reaction likely followed the general mechanism of acetal methanolysis,16 where Brønsted acidity on solid faujasites was the catalyst.17
:GlyF = 30), the reaction of GlyF and DMC was carried out using different molar ratios K2CO3:GlyF (Q) of 0.5, 2, and 3, respectively. Experiments were run for 10 h. Fig. 2 shows the composition (%, by GC) of the reaction mixtures and the O-methylation selectivity. The figure also includes the results obtained with Q = 1.2 (Fig. 2).
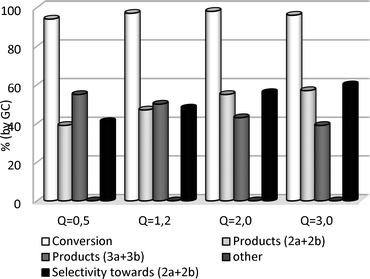 | ||
| Fig. 2 Effect of the K2CO3 amount in the reaction of GlyF with DMC. Q is the molar ratio K2CO3:GlyF. | ||
The conversion of GlyF remained above 94%, even at a Q ratio of 0.5, indicating that the reaction was a truly catalytic process. The base amount however, affected the distribution of O-methyl and carboxymethyl derivatives: as the Q ratio increased (from 0.5 to 3.0), the quantity of products (2a+2b) and (3a+3b) steadily increased and decreased, respectively, from 39 to 57% (light gray bar) and from 55 to 39% (gray bar). Accordingly, the O-methyl selectivity went up from 41 to 60% (black bar). Although the relative increments/decrements of compounds 2 and 3 tended to level off at high catalyst loadings (2.0 ≤ Q ≤ 3.0), the trend agreed with that of Fig. 1: as the reaction proceeded, O-alkyl derivatives were the ultimate products, while carboxymethyl species progressively decreased.
:GlyF = 30; molar ratio K2CO3:GlyF = 1.2); though, a longer reaction time of 20 h was chosen to possibly maximise the formation of products 2. Results are reported in Fig. 3.
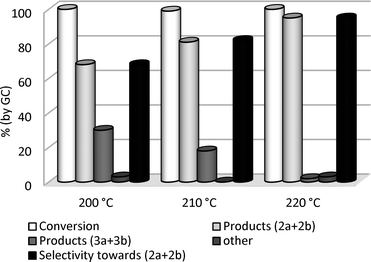 | ||
| Fig. 3 Reactions of GlyF with DMC at different temperatures. | ||
The increase of the temperature induced a remarkable improvement of the O-methylation selectivity (up to 95%, black bar): from 200 to 220 °C, the amount of 2a+2b (light gray bar) increased progressively from 68 to 96%, while carboxymethyl derivatives (3a+3b, gray bar) decreased from 30% to 2%. Further to that, high reaction temperatures did not favour other by-products (≤3% in total). The conversion was substantially quantitative in all cases.
:GlyF (W) of 20, 30, and 40 respectively. According to Fig. 3, the temperature was set to 220 °C. However, experiments were run for 10 h to appreciate possible effects on the product distribution. The molar ratio K2CO3:GlyF was 1.2. Fig. 4 reports the composition (%, by GC) of reaction mixtures and the corresponding O-methylation selectivity.
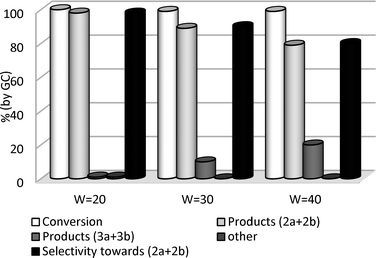 | ||
| Fig. 4 Effect of DMC loading in the reaction of GlyF and DMC. | ||
Surprisingly, the increase of the W ratio from 20 to 40 brought about a remarkable decrease of the selectivity from 98 to 80% (black bar) due to unconverted carboxymethyl derivatives 3: the alkylation of GlyF was apparently disfavoured by a larger excess of DMC.
:GlyF = 30), K2CO3 (molar ratio Q = K2CO3:GlyF = 1.2) was set to react in the presence of n-dodecane (C12; 0.175 mL, 0.131 g, molar ratio, Z, GlyF:C12 = 10) as an internal standard. Conditions were those of Fig. 3 (220 °C, 20 h). The analysis of the final reaction mixture gave conversion and selectivity towards O-methyl derivatives (2a+2b) of 98% and 95%, respectively. The calculated yield of (2a+2b) was 95%. No significant mass loss took place during the reaction.
The reaction of GlyF with DMC was then scaled up. At 220 °C, two experiments (A and B) were performed multiplying by a factor of 5 the amounts of reagents and catalyst. In the first reaction (A), conditions were those of Fig. 3 (W = 30 and Q = 1.2), while in the second test (B), the DMC volume was significantly decreased to reach a W ratio (molar ratio DMC:GlyF) of 5. Accordingly, a mixture of glycerol formal (4.0 g, 38 mmol) and K2CO3 (6.3 g, 46 mmol) was set to react with 96 and 17 mL of DMC, respectively (A and B).
Under the conditions of experiment (B), a further reaction was carried out in the presence of n-dodecane (0.91 mL, molar ratio GlyF:C12 = 10) as the internal standard. This experiment was repeated three times: calculated conversions and product distributions did not differ more than 5% from one test to another. Also, the yield of O-methyl products 2a+2b was determined. Results are summarized in Table 1.
| Entry |
W
a (mol:mol) |
t (h) | IS b (n-C12) | Conv. (%)c | Selectivity (%)c | Yield (2,%)c | |
|---|---|---|---|---|---|---|---|
| (by GC) | Isolatedd | ||||||
|
a Molar ratio DMC:GlyF.
b
IS: internal standard (n-dodecane).
c Conversion of GlyF and selectivity/yield of O-methyl products 2a+2b. Entry 3: values of both conversion, selectivity and yield were averaged over three different runs and they were calculated based on GC calibration curves.
d Isolated yield of distilled products 2a+2b.
|
|||||||
| 1 | 30 | 52 | None | 99 | 94 | 65 | |
| 2 | 5 | 20 | None | 98 | 96 | 64 | |
| 3 | 5 | 20 | Yes | 99 | 97 | 99 | 67 |
The reaction proved to be feasible on a multigram scale. In analogy to results of Fig. 1b, C6 isomers of compounds 2 and 3 were more reactive than the corresponding C5 isomers: for example, after 26 h at 220 °C, the amounts of 2a and 2b were 20 and 41% respectively, while those of 3a and 3b were 27 and 5% (conditions of entry 1, Table 1). However, once the reaction was complete, O-methyl derivatives (2a+2b) were always obtained with a high selectivity (up to 97%) and an excellent yield (99%, by GC).
The alkylation rate was considerably altered by the DMC volume: reaction times were of 52 and 20 h at W = 30 and 5, respectively (entries 1 and 2–3). Apparently, a lower amount of DMC (W = 5) had not only the positive effect of reducing the mass index of the process, but it allowed an even faster reaction thereby confirming the trend of Fig. 4.
Final mixtures from experiments of entries 1–3 of Table 1, were filtered and O-methylation products 2 were distilled under vacuum (further details are in the experimental section). Pure (≥96%) liquid mixtures of isomers 2a and 2b were obtained, though isolated yields did not exceed 67%. Technical limitations of the available distillation apparatus always brought to middle fractions containing the work-up solvent, DMC, and not negligible amounts of products 2.
 | ||
| Scheme 5 | ||
Since conditions for such a process encompassed those of Fig. 1–4, a decarboxylation was expected in the reaction of GlyF with DMC, involving not only DMC, but also non symmetrical carbonates such as carboxymethyl isomers 3a and 3b (Scheme 4). Bearing this in mind, a more in-depth analysis of this aspect was undertaken.
Initially, the decarboxylation of DMC was investigated. Experiments were carried out under the conditions of Fig. 3. In an autoclave, a mixture of DMC (19 mL, 0.23 mol) and K2CO3 (1.28 g, 9.4 mmol, the molar ratio, W1, DMC:K2CO3 was 24), was set to react at temperatures of 200, 210 and 220 °C, and for 20 h. Once the reactor was cooled to rt, a residual pressure up to 5 bar indicated the formation of gaseous products. GC-MS analyses of such gas-phase confirmed the presence of CO2 and dimethyl ether (DME) (Scheme 5). These compounds were recovered by a cold-trap apparatus,19a and weighed: their total amounts were 0.36, 1.26, 2.43 g, at 200, 210, 220 °C, respectively. According to the stoichiometry of Scheme 5, the corresponding conversions of DMC were 2, 6, and 12%. Although of minor entity, the decarboxylation of DMC took place under the conditions used for the methylation of GlyF. The analysis of the residual DMC in the autoclave showed traces of methanol (not exceeding 2%, by GC), indicating that the starting carbonate was also subjected to hydrolysis.20
Then, the reactivity of isomers 3a–3b was examined. Compounds 3 were prepared from GlyF and DMC according to a conventional method for base-catalysed transesterification reactions.21 The products were isolated as a mixture of 3a and 3b (in the same 2:3 ratio of the starting isomers of GlyF) in a 79% yield. They were fully characterized by 1H and 13C NMR, and GC-MS.
The reaction of 3a–3b was performed under the conditions of previous experiments (Fig. 1–4), by replacing DMC with dimethoxyethane (glyme) as the reaction solvent.22 In particular, a mixture of 3a–3b (1.25 g, 7.7 mmol), K2CO3 (1.28 g, molar ratio Q1 = K2CO3:3a–3b = 1.2) and glyme (19 mL) was set to react at 220 °C, in an autoclave, for 10 h. Once cooled to rt, a residual pressure up to 3 bar inside the reactor, indicated that gaseous products were originated. These were recovered and analysed by GC-MS: the exclusive formation of CO2 was observed (details are in the experimental section). This proved unambiguously that, alike to DMC, compounds 3 underwent a decarboxylation reaction. In addition, the (weighed) amount of CO2 (0.20 g) allowed to estimate a 63% conversion of carboxymethyl reagents (3a–3b). The result was in good agreement with the GC analysis of the residual liquid mixture in the autoclave, by which the conversion of 3 resulted 73%. However, the analysis of the liquid phase also indicated that the expected methyl derivatives (2a–2b) were present in a minor amounts [8%, Scheme 6, path (a)], while major products were GlyF isomers (65%). The presence of water in the reaction mixture likely accounted for this behaviour [Scheme 6, path (b)].
 | ||
| Scheme 6 Decarboxylation of the carboxymethyl derivatives (3a–3b) over K2CO3. | ||
To reduce/eliminate the competitive hydrolysis of compounds 3, an additional experiment was carried out using anhydrous glyme and K2CO3. The latter was dried under vacuum (70 °C, 18 mm, overnight). The reaction was performed under the conditions of Scheme 6. However, modest variations were observed: the conversion was 70%, and the amounts of O-methyl products (2a–2b) and GlyF isomers were 19 and 51%, respectively. Overall, the decarboxylation of compounds 3a–3b occurred, but this reaction took place along with a competitive hydrolysis of reactants themselves.
The reaction of solketal with DMC in the presence of K2CO3
The reaction of solketal with DMC was studied based on the results previously obtained for GlyF. Initial reactions were performed in a stainless steel autoclave (200 mL) charged with commercial solketal (1c: 1.02 g; 7.7 mmol), dimethyl carbonate (DMC, 19 mL, molar ratio, W, DMC:1c = 30) and K2CO3 (molar ratio, Q, K2CO3:1c = 1.2). Five experiments were carried out at 220 °C for 20, 30, 50, 60, and 80 h, respectively. Major products (≥95% by GC-MS) were from O-methylation and carboxymethylation reactions of solketal (Scheme 7: compounds 2c and 3c, respectively).
 | ||
| Scheme 7 Major products of the reaction of solketal with DMC catalysed by K2CO3. | ||
These compounds were isolated and fully characterized by GC-MS, 1H and 13C NMR (further details are in the experimental section). Other unidentified by-products whose total amount was ≤5% (by GC), were also detected. The results are reported in Fig. 5.
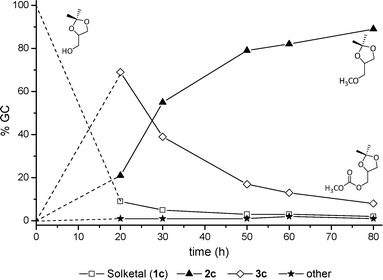 | ||
| Fig. 5 The reaction of solketal with DMC carried out at 220 °C. | ||
As for the case of GlyF, the evaluation of the mass balance and the scale up of the methylation of solketal with DMC were investigated. Results are reported in Table 2.
| Entry | Solketal 1c (g) | DMC:1c (W, mol:mol) a |
T/°C | t (h) | IS b (n-C12) | Conv. (%)c | Sel. (%)c | Yield (2c,%) |
|---|---|---|---|---|---|---|---|---|
| The K2CO3:1c molar ratio was 1.2.a Molar ratio DMC:GlyF.b IS: internal standard (n-dodecane, C12). The 1c:C12 molar ratio was 10.c Conversion of solketal and selectivity/yield of O-methyl product 2c. Entry 1: values of both conversion, selectivity and yield were calculated based on GC calibration curves.d Isolated yield of 2c. |
||||||||
| 1 | 1.02 | 30 | 220 | 80 | Yes | 95 | 92 | 86c |
| 2 | 3.50 | 5 | 50 | None | 92 | 99 | 66d |
The mass balance was examined in the presence of n-dodecane as internal standard (entry 1). Reaction conditions were those of Fig. 5 (80 h). Calculated conversion, O-methyl selectivity, and yield on 2c were 95, 92 and 86%, respectively. With respect to solketal, these values confirmed that no significant mass loss took place during the reaction.
The reaction was then scaled up. With respect to conditions of Fig. 5, the amounts of solketal and the catalysts were multiplied by a factor of 3.5. Instead, the volume of DMC was adjusted (lowered) to reach a molar ratio DMC:solketal = 5. After 50 h, conversion and O-methyl selectivity were excellent (92 and 99%, respectively; entry 2), thereby proving the feasibility of the reaction on a multigram scale. The final mixture appeared as a thick slurry whose analysis showed that only minor amounts of DMC (20% with respect to product 2c) were still present. Not only the occurrence of the decarboxylation/hydrolysis of dimethyl carbonate was confirmed, but the process became rather extensive at long reaction times.
The mixture was diluted with diethyl ether and filtered, and the O-methylation product 2c was distilled under vacuum (56 °C, 3 mmHg) as a pure (98%, by GC) colourless liquid. The isolated yield of 2c was of 2.50 g (66%). As for GlyF, the available distillation apparatus brought also to middle fractions containing the work-up solvent, DMC and MeOH, and not negligible amounts of 2c.
The K2CO3-catalyzed reaction of solketal with dibenzyl- and diethyl-carbonate
In order to expand the scope of the alkylation of glycerol acetals with dialkyl carbonates, the reaction of solketal with both dibenzyl- and diethyl-carbonate (DBnC and DEC, respectively) was investigated.23 Experiments were carried out under conditions similar to those used for DMC. In a stainless steel autoclave, a mixture of solketal (1c, 1.8 mL, 14.6 mmol), K2CO3 (2.4 g, molar ratio, Q, K2CO3:1c = 1.2) and different amounts of DBnC or DEC (molar ratios, W, dialkyl carbonate:1c = 5 and 8 were used) was set to react in the range of 190–220 °C, for 18–66 h. Final reaction mixtures were analysed by GC-MS. Both products of O-alkylation and carboxyalkylation of solketal were observed (Scheme 8: derivatives 4c–5c, and 6c–7c, respectively).
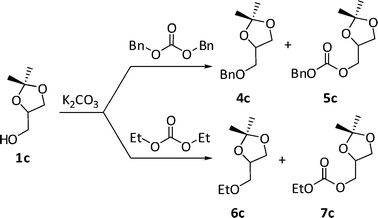 | ||
| Scheme 8 Major products of the K2CO3-catalysed reaction of solketal with DBnC and DEC. | ||
Compounds 4c and 5c were isolated and characterized by GC-MS, 1H and 13C NMR, while the structures of products 6c and 7c were assigned from their mass spectra. Results are reported in Table 3.
| Entry | DAlC |
DAlC:1c, W (mol:mol) |
T/°C | t (h) | Conv. (%)a | O-alkyl selectivity (%) | Products (%, by GC)b | Yield (%)c | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 4c | 5c | 6c | 7c | Others | ||||||||
| a Conversion of solketal. b 4c and 6c: O-benzyl and O-ethyl derivatives of solketal, respectively. 5c and 7c: O-carboxybenzyl and O-carboxyethyl derivatives of solketal, respectively. c Isolated yield of product 4c. | ||||||||||||
| 1 | DBnC | 5 | 190 | 18 | 85 | 22 | 19 | 62 | 4 | |||
| 2 | 8 | 83 | 8 | 7 | 71 | 5 | ||||||
| 3 | 5 | 220 | 18 | 78 | 97 | 76 | 1 | 1 | ||||
| 4 | 8 | 18 | 80 | 85 | 68 | 12 | ||||||
| 5 | 8 | 32 | 81 | 100 | 81 | 80 | ||||||
| 6 | DEC | 5 | 220 | 18 | 80 | 1 | 1 | 76 | 3 | |||
| 7 | 66 | 54 | 100 | 54 | ||||||||
The increase of the temperature from 190 to 220 °C boosted the formation of product 4c: the O-benzyl selectivity rose from 8–22% to 97% (entries 1–2 and 3–4). Contrary to our expectations, the conversion of solketal remained rather constant in all experiments (around 80%, entries 1–4).
This incongruity was plausibly due to: i) a partial hydrolysis of the carboxybenzyl product 5c which brought back to the starting reactant 1c (as in Scheme 6); ii) extensive decarboxylation/hydrolysis reactions of DBnC. Processes (ii) were confirmed by the analysis of the final reaction mixture: at 220 °C, after 18 h (W = 8), DBnC was absent and sizeable amounts of dibenzyl ether and benzyl alcohol (41 and 46%, respectively, with respect to O-methyl product 4c, 13%) were detected (entry 4).24 (Scheme 9).
Under the same conditions (220 °C, W = 8), an additional reaction carried out for a prolonged time (32 h) proceeded with total selectivity towards compound 4c, but the conversion of solketal did not exceed 81% (entry 5). Again, not traces of DBnC were detected in the final mixture.
Discussion
The mechanism of DMC-mediated methylations of GlyF and solketal
In the presence of K2CO3 as a catalyst, the reactions of dimethyl carbonate with glycerol formal (GlyF) and with solketal show remarkable analogies. First of all, experimental conditions can be found for the formation of O-methyl ethers of both substrates with a very high selectivity (>90%) at complete conversion (Fig. 3 and 5, Tables 1 and 2). This unexpected and never previously reported result fulfils the synthetic goal of the present work. It should be noted that under basic catalysis, alcohols are claimed to react with dialkyl carbonates to form transesterification and not alkylation products (Scheme 2). Other important similarities between the methylation of GlyF and of solketal emerge when the effects of the temperature (T) and of the reaction time (t) on the product distribution are considered (Fig. 1, 3, and 5).Both alkylations of GlyF and solketal yield O-methyl and carboxymethyl derivatives (2 and 3, respectively); however, as T and/or t are increased, the amount of carboxymethyl compounds rises to a maximum and then, it drops to zero. At the same time, O-methyl compounds always increase (though at different rates with respect to 3) to become final reaction products.
Two plausible explanations can be offered for the formation/disappearance of compounds 3: i) these products are originated by a reversible reaction (transesterification), i.e. an addition/elimination at the DMC carbonyl;25ii) compounds 3a–3b (from GlyF) are consumed by competitive decarboxylation/hydrolysis processes which are triggered by temperature (Fig. 3 and Scheme 6).26
Based on these observations and on the sequence for base-catalyzed DMC-mediated reactions,10,11Scheme 10 illustrates a mechanistic hypothesis for the O-methylation of GlyF and solketal with DMC.
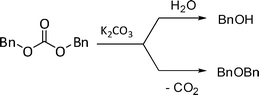 | ||
| Scheme 9 Hydrolysis and decarboxylation of DBnC. | ||
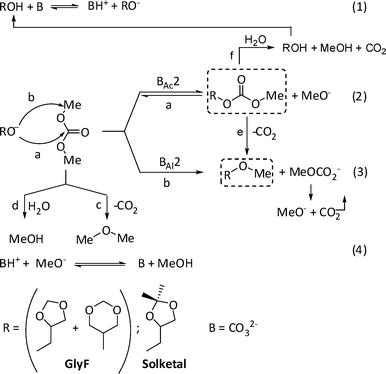 | ||
| Scheme 10 The mechanism for the methylation of GlyF and solketal with DMC. | ||
In the first step, the base (B) converts the acetal/ketal (ROH: GlyF or solketal) in the corresponding alcoholate anion [RO−, eqn (1)] that reacts with both electrophilic centers of DMC [paths a and b, BAc2 and BAl2 mechanisms of eqn (2) and (3)]. Since the carbonyl carbon is a stronger electrophilic site than the methyl one, a lower activation energy is expected for the transesterification [path (a)] with respect to the methylation reaction [path (b)]. This may account for the faster formation of carboxymethyl- (3: ROCO2Me) with respect to methyl- (2: ROMe) derivatives (Fig. 1 and 5). However, the onset of the methylation process makes the reversible transesterification backtrack: after an initial accumulation of compounds 3, they decrease to zero, while ROMe become final products.
A further contribution to the disappearance of carboxymethyl products is given by competitive decarboxylation and hydrolysis reactions,20 shown in paths (e) and (f), respectively. For a complete view, also the decarboxylation/hydrolysis of DMC is indicated [paths (c)–(d)]: the extent of such transformations become relevant only for long reaction times.
The catalytic cycle terminates when the protonated base (BH+) reacts with an alcoholate anion (e.g.MeO−) to restore the free base (B) and produce MeOH [eqn (4)].
Overall, several concurrent processes take place, but if temperature is high enough and sufficient time is allowed, the system equilibrates to thermodynamically favoured products: the result is the selective formation of O-methyl derivatives of both GlyF and solketal.
In the case of GlyF, the higher rate of formation/disappearance of 6-membered ring isomers (2b and 3b) with respect to their 5-membered ring analogues (2a and 3a) is an additional aspect (Fig. 1b and Table 1). Despite the huge literature on glycerol formal, very few papers describe fundamental studies on the behaviour of isomers of glycerol acetals and ketals. Among them, a work by Aksnes et al. has investigated 5- and 6-membered ring acetals obtained by the reaction of glycerol with acetaldehyde (Scheme 11: compounds a and b, respectively).27
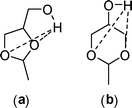 | ||
| Scheme 11 (a) 4-Hydroxymethyl-2-methyl-1,3-dioxolane; (b) 5-hydroxy-2-methyl-1,3-dioxane. Dashed lines: intramolecular H-bonds. | ||
Authors provide evidence for intramolecular H-bonds (dashed lines of Scheme 11) between the hydroxyl proton and endocyclic oxygen atoms of both acetals (a) and (b). Moreover, stereochemical reasons particularly, the mutual interactions between axial hydrogen atoms, offer an explanation for the different stability of (a) with respect to (b). This seems a promising basis also for the investigated reaction even though our experimental conditions, above all the high temperature (≥200 °C), plausibly tend to smooth energetic differences due to hydrogen bonds or to conformational barriers. Further studies will be devoted to a more in-depth analysis of such aspects.
The role of the catalyst (K2CO3)
Fig. 2 shows that the reaction of GlyF with DMC takes place even when a substoichiometric amount of K2CO3 is used (Q = 0.5). Increase of the catalyst loading speeds up the reaction until a sort of plateau is reached: at Q ≥ 2, further improvements of rate and/or selectivity are less, if at all, appreciable. Although a kinetic analysis is beyond the scope of this work, it should be considered that K2CO3 is only moderately soluble in DMC (0.58 g L−1 at 180 °C,11a). Therefore, the investigated reaction is plausibly partitioned between a liquid homogeneous phase saturated with the base and the solid catalytic surface where organic reactants/products may adsorb/desorb. As the Q ratio increases, the homogeneous contribution should remain constant, while the heterogeneous reaction over K2CO3 should be favored until the solid surface accommodates the maximum allowed quantity of organic substrates. After that, the reaction rate cannot increase any further and the selectivity levels off as well.A similar behavior has been observed also for other base-catalysed reactions of DMC, for example in the methylation of CH2-active compounds.28
The DMC amount
Fig. 4 and Tables 1–2 indicate that the decrease of DMC:substrate molar ratio (W) favors the alkylation of both GlyF and solketal. This is particularly evident in scale-up experiments which show that the reaction time halves when the W ratio is reduced from 30 to 5. The reasons of this behaviour are not clear, but some speculations can be made. A dilution effect is hardly responsible for the result since DMC is also a reactant. However: i) at W = 5, the high concentration of GlyF or solketal may favour the base dissolution. It should be noted that the solubility of K2CO3 boosts by a factor of 22 (from 0.58 to 12.7 g L−1) when DMC is compared to methanol,29 and both acetals (GlyF or solketal) possess polar alcohol-like structures. An increase of the reaction rate is plausible if the catalyst solubility is improved. Moreover, as the reaction proceeds, GlyF (or solketal) is gradually converted to the corresponding methyl and carboxymethyl derivatives, but an equivalent amount of methanol is originated to restore a medium polarity similar to that of the starting solution. The effect of decarboxylation/hydrolysis of DMC may also play a role, especially for prolonged experiments (above 50 h), where such transformations occur to a sizeable extent. ii) As mentioned above, the investigated reaction is likely to occur both in solution and over solid K2CO3. For a number of reasons including solvation effects, competitive adsorption, alteration of diffusion constants, etc., the modes of (physical/chemical) interactions of reagents (GlyF/solketal and DMC) on the catalytic surface as well as the regime of the reaction can be modified by varying the volume of the solvent (DMC).30
The K2CO3-catalyzed decarboxylation of DMC and carboxymethyl derivatives 3
We have recently reported that a class of zeolites (alkali metal exchanged faujasites) are efficient catalysts for the decarboxylation of dialkyl carbonates.19a The more basic the zeolite (NaX, CsY), the higher its activity. This feature has been explained by the excellent potential for CO2 capture/storage of these catalysts that are able to adsorb reversibly up to 5.5 mmol of CO2 per g of solid.31 Accordingly, the breakdown of dialkyl carbonates is favoured and, at the same time, the reversibility of the CO2 uptake prevents the saturation of the catalytic surface by the gas. This leaves a sufficient solid basicity to allow the decarboxylation to proceed. An similar interpretation may be offered for the activity of K2CO3 in the decarboxylation reactions of both DMC and compounds 3a–3b and 3c (Schemes 5–6 and Fig. 5). In fact, K2CO3 as such or supported on various porous materials (C or silica gel), is a very good CO2 sorbent system on condition that water acts as a co-reagent (Scheme 12).32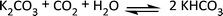 | ||
| Scheme 12 The chemical adsorption of CO2 over K2CO3. | ||
This process may take place under the conditions described in this work where water is plausibly present due to hygroscopic properties of K2CO3 or the reactants.33 Whatever its origin, the presence of water in our system is witnessed by the extensive hydrolysis observed for carbonates 3a–3b (Scheme 6) and DBnC as well. It should be finally noted that the solid basicity per se is not a sufficient requisite for a rapid decarboxylation reaction: solid K2CO3 is by far a poorer catalyst with respect to amphoteric faujasites. A possible reason lies in a inefficient adsorption of dialkyl carbonates on K2CO3 with respect to zeolites.34
Other dialkyl carbonates
Previous studies by our group have demonstrated that in the alkylation of several nucleophiles (including CH2-active compounds and amines), dibenzyl carbonate (DBnC) displays a reactivity comparable if not higher than DMC;35 while, diethyl carbonate (DEC) is a less active alkylation agent than DMC.36 The same behavior holds true for the alkylation of solketal investigated here (compare Tables 2 and 3). Moreover, although the study of such a reaction is at a preliminary stage, the formation of O-alkyl and carboxyalkyl compounds (4c–6c, and 5c–7c, respectively) from DBnC and DEC, is plausibly explained by a mechanism analogue to that outlined in Scheme 10 for DMC. Table 3 also shows that excellent O-alkyl selectivities (up to 100%) are achieved; in all cases, notwithstanding the excess of dialkyl carbonates, conversions are never quantitative and they level off, if not decrease, at high temperature and for prolonged reactions. As for DMC, the occurrence of decarboxylation/hydrolysis processes decompose the starting carbonates: results indicate that DBnC is a more efficient alkylating agent, but it also disappears at a higher rate with respect to DEC. Resonance effects similar to those invoked for SN2 displacements likely enhance the reactivity of the benzyl system.37Conclusions
The set up of innovative transformations of glycerol and its derivatives represents an appealing challenge of modern chemistry. Not only for the fact that glycerol is among the most valuable biomass derived platform chemicals, but also for the market surplus of glycerol due to biodiesel productions. This work demonstrates that in the presence of K2CO3 as a base catalyst, dimethyl carbonate reacts with acetals of glycerol such as glycerol formal and solketal, to give the selective formation of the corresponding methyl ethers: at complete conversion, these products are obtained with selectivity up to 99%. These never previously reported reactions can be also scaled up for multigram synthesis without loss of selectivity; interestingly, under such conditions, the use of a reduced volume of DMC allows a consistent decrease of the reaction time and of the mass index of the process. Although the reaction is rather energy intensive, the procedure has a genuine potential for its low environmental impact. It is a catalytic process that couples the use of non-toxic dimethyl carbonate with renewable-derived acetals/ketals. Also, no additional solvents are required and major by-products are methanol and CO2. An accurate analysis of the mass balance indicates that excellent GC yields (>90%) of O-methylation products (2) can be achieved, meaning that no mass loss occurs with respect to starting acetals. However, due to technical limitations of the purification (distillation) step, compounds 2 are isolated in moderate yields not exceeding 67%.Beyond the methylation process, the overall transformation includes the transesterification of both GlyF and solketal with DMC to produce the corresponding carboxymethyl derivatives. These compounds follow an intermediate-like behaviour which makes them disappear as the reaction proceeds: the reversible nature of the transesterification and the decarboxylation/hydrolysis of carboxymethyl products account for this result. The latter processes become significant also for DMC when prolonged reactions are performed. To the best of our knowledge, this behavior has never been previously described for reactions of alcohols and dialkyl carbonates under basic catalysis conditions, and it may open interesting synthetic perspectives. Also, a mechanistic hypothesis has been formulated on this basis.
The preliminary investigation of the reaction of solketal with both dibenzyl- and diethyl-carbonate suggests that the alkylation of glycerol acetals with dialkylcarbonates may be of a general scope. However, the extensive decarboxylation/hydrolysis of the starting carbonates does not allow the complete conversion of solketal. To limit side-reactions, studies are in progress with other base catalysts able to operate at lower temperatures than K2CO3.
Experimental
General
Glycerol formal (GlyF, 1a+1b), solketal (1c), benzyl alcohol, dimethyl carbonate (DMC), diethyl carbonate (DEC), dimethoxyethane (glyme), n-dodecane (C12), K2CO3, ethyl acetate (EA), petroleum ether (PE) and diethyl ether (Et2O), were ACS grade from Aldrich. If not otherwise specified, they were employed without further purification. Dibenzyl carbonate (DBnC) was prepared via the transesterification reaction of benzyl alcohol with DMC, by adjusting a method reported in the literature18 (further details are in the ESI†). NaX faujasite was from Aldrich: before its use, it was dehydrated under vacuum (70 °C, 18 mm, overnight). GC-MS (EI, 70 eV) analyses were run using a FFAP capillary column (30 m), and CG/FID analyses were run using a DBWAX capillary column (30 m). 1H NMR were recorded at 400 MHz, 13C NMR spectra at 100 MHz, and chemical shifts were reported in δ values downfield from TMS; CDCl3 was used as solvent.Reaction procedures
:1 = 30) and K2CO3 (1.28 g, molar ratio, Q, K2CO3:1 = 1.2). Where indicated, reactions with internal standard were carried out by adding n-dodecane (0.175 mL, 0.131 g, molar ratio, Z, 1:C12 = 10) to the mixture. The autoclave was purged with N2, electrically heated at the desired temperature (200–220 °C) and kept under magnetic stirring throughout the reaction. At the end of the experiment, the autoclave was cooled to rt and then to −60 °C. The reactor was slowly vented until it returned to rt. The reaction mixture was finally analyzed by GC.
This procedure was adapted to examine the effect of reaction parameters on the alkylation of both GlyF and solketal with DMC: accordingly, the temperature, the time, the catalyst loading and DMC amount were modified as reported in Fig. 1–5 and Table 2.
:GlyF = 30] and K2CO3 [6.3 g, 46 mmol, the molar ratio Q = K2CO3:GlyF = 1.2] was set to react at 220 °C (Table 1). Since a sizeable quantity of CO2 was generated during the reaction, once a pressure of 60 bar was reached (after ∼25 h), the autoclave was rapidly cooled to rt, and then to −60 °C in a liquid nitrogen bath, and slowly vented. After that, the autoclave was re-purged with N2 and re-heated at 220 °C. The release of CO2 overpressure was repeated at 37 and 43 h, respectively. The reaction was finally stopped after 52 h (Table 1, entry 1).
The same procedure was used to scale up the reaction in the presence of: i) a reduced volume DMC (17 mL, molar ratio, W, DMC:GlyF = 5; Table 1, entry 2), and ii) n-dodecane as an internal standard (0.861 mL, 0.646 g, molar ratio 1:C12 = 10; Table 1, entry 3).
The same procedure was used to scale up the reaction of solketal with DMC. In this case, the amounts of solketal and K2CO3 were multiplied by a factor of 3.5, while DMC was used in a5-molar excess over the substrate: a mixture of solketal (1c, 3.5 g, 26 mmol), DMC (11 mL) and K2CO3 [4.3 g, 31 mmol, the molar ratio, Q, K2CO3:1c = 1.2) (Table 2, entry 2) was set to react at 220 °C for 50 h.
:K2CO3 was 24), was set to react for 20 h at temperatures of 200, 210 and 220 °C. In the case of carboxymethyl derivatives 3a–3b, a mixture of isomers 3a+3b (1.25 g, 0.0077 mmol), K2CO3 (1.28 g, molar ratio, Q1, K2CO3:3a–b = 1.2), and dimethoxyethane (glyme, 19 mL) as solvent, was set to react at 220 °C for 10 h. Under these conditions, an additional experiment was carried out using anhydrous solvent and catalyst: glyme was dried through conventional methods,38 while K2CO3 was dried under vacuum (70 °C, 18 mm, overnight).
At the end of experiments, gaseous products generated from decarboxylation reactions, were recovered by a cold-trap apparatus,19a weighed and analysed by GC-MS. In the case of DMC, mixtures of CO2 and dimethyl ether (DME) were obtained:39 the corresponding amounts were 0.36, 1.26 and 2.43 at 200, 210 and 220 °C, respectively. In the case of carboxymethyl derivatives 3a–3b, only CO2 was recovered: the amount was 0.20 and 0.23 g for experiments in non-anhydrous and anhydrous glyme/catalysts, respectively.
Isolation and characterization of O-alkyl products
:3 ratio of the starting isomers of GlyF. Yields of 2a+2b were 2.91, 2.87 and 3.00 g (65, 64 and 67%; Table 1 entries 1, 2, and 3, respectively). Compounds 2a–2b were fully characterized by GC-MS, 1H and 13C NMR.
GC-MS (relative intensity, 70 eV) m/z: 2a 118 (M+, <1%), 117 ([M − H]+, 1), 88 [M-H2CO]+, 15), 73 ([M-H2COCH3]+, 22), 72 (14), 59 (11), 45 ([CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) OCH3]+, 100), 44 (17), 43 (11), 31 (15), 29 (29), 28 (12), 27 (10); 2b 118 ([M]+, 2%), 88 ([M-H2CO]+, 11), 58 ([M-(H2CO)2]+, 100), 43 ([M-(H2CO)2-CH3]+, 34), 31 (14), 29 (30), 28 (20). 1H NMR (CDCl3, 400 MHz) δ (ppm): 2a 5.01 (s, 1H), 4.86 (s, 1H), 4.22–4.15 (m, 1H), 3.94 (dd, 1H, J1 = 8.1 Hz, J2 = 6.9 Hz), 3.66 (dd, 1H, J1 = 8.2 Hz, J2 = 6.0 Hz), 3.47–3.41 (m, 2H), 3.38 (s, 3H); 2b 4.83 (d, 1H, J = 6.1 Hz), 4.73 (d, 1H, J = 6.1 Hz), 4.02 (dd, 2H, J1 = 11.6 Hz, J2 = 3.5 Hz), 3.71 (dd, 2H, J1 = 11.6 Hz, J2 = 6.1 Hz), 3.40 (s, 3H), 3.32–3.26 (m, 1H). 13C NMR (CDCl3, 100 MHz) δ (ppm): 2a 95.3,74.3, 68.9, 66.9, 56.8; 2b 93.6, 72.9, 71.3, 54.8.
OCH3]+, 100), 44 (17), 43 (11), 31 (15), 29 (29), 28 (12), 27 (10); 2b 118 ([M]+, 2%), 88 ([M-H2CO]+, 11), 58 ([M-(H2CO)2]+, 100), 43 ([M-(H2CO)2-CH3]+, 34), 31 (14), 29 (30), 28 (20). 1H NMR (CDCl3, 400 MHz) δ (ppm): 2a 5.01 (s, 1H), 4.86 (s, 1H), 4.22–4.15 (m, 1H), 3.94 (dd, 1H, J1 = 8.1 Hz, J2 = 6.9 Hz), 3.66 (dd, 1H, J1 = 8.2 Hz, J2 = 6.0 Hz), 3.47–3.41 (m, 2H), 3.38 (s, 3H); 2b 4.83 (d, 1H, J = 6.1 Hz), 4.73 (d, 1H, J = 6.1 Hz), 4.02 (dd, 2H, J1 = 11.6 Hz, J2 = 3.5 Hz), 3.71 (dd, 2H, J1 = 11.6 Hz, J2 = 6.1 Hz), 3.40 (s, 3H), 3.32–3.26 (m, 1H). 13C NMR (CDCl3, 100 MHz) δ (ppm): 2a 95.3,74.3, 68.9, 66.9, 56.8; 2b 93.6, 72.9, 71.3, 54.8.
GC-MS (relative intensity, 70 eV) m/z: 146 (M+, <1%), 131 ([M-H3C]+, 26), 101 ([M-H2COCH3]+,4), 71 (24), 59 (8), 58 ([M-H3C-(H2CO)-(H3CCO), 100), 45 ([CH2OCH3]+, 4), 43 ([H3CCO]+, 48), 42 (11), 41 (11). 1H NMR (CDCl3, 400 MHz) δ (ppm): 4.28–4.22 (m, 1H), 4.03 (dd, 1H, J1 = 8.2 Hz, J2 = 6.4 Hz), 3.67 (dd, 1H, J1 = 8.2 Hz, J2 = 6.5 Hz), 3.44 (dd, 1H, J1 = 9.8 Hz, J2 = 6.1 Hz), 3.38 (dd, 1H, J1 = 9.8 Hz, J2 = 5.1 Hz), 3.37 (s, 3H), 1.40 (s, 3H), 1.34 (s, 3H). 13C NMR (CDCl3, 100 MHz) δ (ppm): 109.3, 74.5, 73.7, 66.6, 59.2, 26.6, 25.3.
:1:1 v/v]. Isolated yield of 4c was of 80% (2.58 g). Product 4c was fully characterized by GC-MS, 1H and 13C NMR.
GC-MS (relative intensity, 70 eV) m/z: 222 (M+, <1%), 207 ([M-H3C]+, 12), 105 (11), 101 ([M-H2COCH2Ph]+, 28), 92 (12), 91 ([CH2Ph]+, 100), 65 (11), 43 ([H3CCO]+, 35). 1H NMR (CDCl3, 400 MHz) δ (ppm): 7.40–7.27 (m, 5H), 4.62–4.53 (m, 2H), 4.34–4.26 (m, 1H) 4.05 (dd, 1H, J1 = 8.2 Hz, J2 = 6.4 Hz), 3.74 (dd, 1H, J1 = 8.3 Hz, J2 = 6.3 Hz), 3.55 (dd, 1H, J1 = 9.8 Hz, J2 = 5.7 Hz), 3.47 (dd, 1H, J1 = 9.8 Hz, J2 = 5.5 Hz), 1.42 (s, 3H), 1.36 (s, 3H). 13C NMR (CDCl3, 100 MHz) δ (ppm): 138.0, 128.4, 127.7, 109.4, 74.7, 73.5, 71.1, 66.9, 26.8, 25.4.
Synthesis, isolation and characterization of carboxyalkyl products
Carboxyalkyl products were prepared by adjusting a conventional method for base-catalyzed transesterification reactions.21:GlyF = 20) and K2CO3 (15.9 g, molar ratio, Q, K2CO3:GlyF = 1.2). The flask was immersed in a thermostated oil bath, heated to the reflux temperature of DMC (90 °C) and kept under magnetic stirring. The reaction was monitored by GC. After 55 h, the mixture was cooled to rt, and filtered to remove K2CO3 (Gooch#4). The solid was washed with diethyl ether (3 × 30 mL). The resulting solution was concentrated by rotary evaporation (30 °C, 600 mbar), and then vacuum distilled. Compounds 3a and 3b were isolated (65 °C, 3 mmHg) as a mixture of isomers in approximately the same ratio of the starting isomers of GlyF. Yield of 3a–3b was 79% (12 g, purity 98%, by GC). The products were fully characterized by GC-MS, 1H and 13C NMR.
GC-MS (relative intensity, 70 eV) m/z: 3a 162 (M+, <1%), 161 ([M − H]+, 4), 103 ([M-CO2CH3]+, 14), 102 (34), 86 ([M-H-OCO2CH3]+, 49), 77 (14), 73 ([M-CH2OCO2CH3]+, 44), 59 ([CH3OC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) O]+, 40), 58 (43), 57 (38), 55 (11), 45 ([CHOCH3]+, 100), 44 (36), 43 (50), 31 (52), 30 (10), 29 ([CHO]+, 98), 28 (34), 27 (25); 3b 162 (M+, <1%), 161 ([M − H]+, 4%), 102 ([M-(H2CO)2]+, 72), 86 ([M-H-CO2-OCH3]+, 49), 59 ([CH3OCO]+, 41), 58 ([M-(H2CO)2-CO2]+, 65), 57 (33), 55 (16), 45 (63), 44 (21), 43 ([M-(H2CO)2-CO2CH3]+, 72), 31 (59), 30 (14), 29 ([CHO]+,100), 28 (44), 27 (27). 1H NMR (CDCl3, 400 MHz) δ (ppm): 3a 5.03 (s, 1H), 4.89 (s, 1H), 4.32–4.25 (m, 1H), 4.21 (dd, 1H, J1 = 11.3 Hz, J2 = 5.7 Hz), 4.16 (dd, 1H, J1 = 11.3 Hz, J2 = 5.2 Hz), 3.97 (m, 1H), 3.79 (s, 3H), 3.72 (dd, 1H, J1 = 8.5 Hz, J2 = 5.5 Hz); 3b 4.89 (d, 1H, J = 6.3 Hz), 4.81 (d, 1H, J = 6.2 Hz), 4.60–4.57 (m, 1H), 4.03 (dd, 2H, J1 = 12.2 Hz, J2 = 2.9 Hz), 3.96 (dd, 2H, J1 = 12.1 Hz, J2 = 4.2 Hz). 13C NMR (CDCl3, 100 MHz) δ (ppm): 3a+3b 155.5, 155.1, 95.5, 95.4, 72.5, 67.2, 66.6, 54.9, 54.9.
O]+, 40), 58 (43), 57 (38), 55 (11), 45 ([CHOCH3]+, 100), 44 (36), 43 (50), 31 (52), 30 (10), 29 ([CHO]+, 98), 28 (34), 27 (25); 3b 162 (M+, <1%), 161 ([M − H]+, 4%), 102 ([M-(H2CO)2]+, 72), 86 ([M-H-CO2-OCH3]+, 49), 59 ([CH3OCO]+, 41), 58 ([M-(H2CO)2-CO2]+, 65), 57 (33), 55 (16), 45 (63), 44 (21), 43 ([M-(H2CO)2-CO2CH3]+, 72), 31 (59), 30 (14), 29 ([CHO]+,100), 28 (44), 27 (27). 1H NMR (CDCl3, 400 MHz) δ (ppm): 3a 5.03 (s, 1H), 4.89 (s, 1H), 4.32–4.25 (m, 1H), 4.21 (dd, 1H, J1 = 11.3 Hz, J2 = 5.7 Hz), 4.16 (dd, 1H, J1 = 11.3 Hz, J2 = 5.2 Hz), 3.97 (m, 1H), 3.79 (s, 3H), 3.72 (dd, 1H, J1 = 8.5 Hz, J2 = 5.5 Hz); 3b 4.89 (d, 1H, J = 6.3 Hz), 4.81 (d, 1H, J = 6.2 Hz), 4.60–4.57 (m, 1H), 4.03 (dd, 2H, J1 = 12.2 Hz, J2 = 2.9 Hz), 3.96 (dd, 2H, J1 = 12.1 Hz, J2 = 4.2 Hz). 13C NMR (CDCl3, 100 MHz) δ (ppm): 3a+3b 155.5, 155.1, 95.5, 95.4, 72.5, 67.2, 66.6, 54.9, 54.9.
:1c = 20) and K2CO3 (3.9 g, molar ratio, Q, K2CO3:1c = 1.2). The flask was immersed in a thermostated oil bath, heated to the reflux temperature of DMC (90 °C) and kept under magnetic stirring. The reaction was monitored by GC. After 95 h, the mixture was cooled to rt, and filtered to remove K2CO3 (Gooch#4). The solid was washed with diethyl ether (3 × 30 mL). The resulting solution (∼100 mL) was concentrated by rotary evaporation (30 °C, 600 mbar), and then vacuum distilled. Product 3c was isolated by distillation under reduced pressure (63 °C, 3 mmHg), in 88% yield (3.85 g, purity 98% by GC). The product was fully characterized by GC-MS, 1H and 13C NMR.
GC-MS (relative intensity, 70 eV) m/z: 190 (M+, <1%), 175 ([M-H3C]+,18), 115 (M-OCO2CH3]+, 2), 101 (M-CH2OCO2CH3]+, 8), 71 (19), 59 ([CH3OCO]+, 16), 57 (11), 43 ([H3CCO]+, 100), 41 (17). 1H NMR (CDCl3, 400 MHz) δ (ppm): 4.36–4.31 (m, 1H), 4.19–4.15 (m, 2H), 4.08 (dd, 1H, J1 = 8.6 Hz, J2 = 6.4 Hz), 3.78 (dd, 1H, J1 = 8.6 Hz, J2 = 5.8 Hz), 3.79 (s, 3H), 1.43 (s, 3H), 1.36 (s, 3H). 13C NMR (CDCl3, 100 MHz) δ (ppm): 155.5, 190.8, 73.2, 67.9, 66.2, 54.9, 26.6, 25.3.
:1c = 2), K2CO3 (1.3 g, molar ratio, Q, K2CO3:1c = 1.2) and dimethoxyethane (glyme, 20 mL) as the solvent. The flask was immersed in a thermostated oil bath, heated to the reflux temperature of glyme (85 °C) and kept under magnetic stirring. The reaction was monitored by GC-MS. After 12 h, the mixture was cooled to rt, and filtered to remove K2CO3 (Gooch#4). The solid was washed with diethyl ether (3 × 10 mL). Compound 5c was purified by FCC (flash column chromatography) over silica gel, using gradient petroleum ether (PE)/diethyl ether (Et2O) elutions [initial PE:Et2O = 1:0 v/v, final PE:Et2O = 1:1 v/v], and isolated in a yield of 65% (1.2 g). Product was fully characterized by GC-MS, 1H and 13C NMR.
GC-MS (relative intensity, 70 eV) m/z: 266 (M+, <1%), 101 ([M-H2COCO2CH2Ph]+,11), 91 ([CH2Ph]+, 100), 65 (10), 59 (10), 43 ([H3CCO]+, 64). 1H NMR (CDCl3, 400 MHz) δ (ppm): 7.39–7.33 (m, 5H), 5.17 (s, 2H), 4.35–4.31 (m, 1H), 4.21 (dd, 1H, J1 = 9.2 Hz, J2 = 3.2 Hz), 4.16 (dd, 1H, J1 = 9.2 Hz, J2 = 3.9 Hz), 4.07 (dd, 1H, J1 = 8.5 Hz, J2 = 6.4 Hz), 3.78 (dd, 1H, J1 = 8.5 Hz, J2 = 5.9 Hz), 1.42 (s, 3H), 1.36 (s, 3H). 13C NMR (CDCl3, 100 MHz) δ (ppm): 149.7, 129.8, 123.4, 123.2, 104.7, 68.1, 64.6, 62.7, 61.1, 21.4, 20.1.
Acknowledgements
MIUR (Italian Ministry for Education, University and Research) is gratefully acknowledged for the financial support of this work.Notes and references
- (a) Z. Z. J. Wang, J. Zhuge, H. Fang and A. Prior, Biotechnol. Adv., 2001, 19, 201–223 CrossRef CAS; (b) J. Chowdury and K. Fouky, Chem. Eng., 1993, 100, 35–39 Search PubMed; (c) C. W. Chiu, M. J. Goff and G. J. Supples, AIChE J., 2005, 51, 1274–1278 CrossRef CAS; (d) F. Jérôme, Y. Pouilloux and J. Barrault, ChemSusChem, 2008, 1, 586–613 CrossRef CAS; (e) M. Pagliaro and M. Rossi, The Future of Glycerol: New Usages for a Versatile Raw Material, 2008, RSC Publishing, Cambridge, UK Search PubMed.
- T. Werpy and G. Petersen, in Top Value Added Chemicals From Biomass, the Pacific Northwest National Laboratory (PNNL) and the National Renewable Energy Laboratory (NREL), U.S. Department of Energy, 2004 Search PubMed.
- (a) G. Vicente, J. A. Melero, G. Morale, M. Paniagua and E. Martin, Green Chem., 2010, 12, 899–907 RSC; (b) European Biodiesel Board, 2003–2009. http://www.ebb-eu.org.
- (a) MSDS at http://sigmaaldrich.com; (b) J. L. Gras, R. Nouguier and M. Mchich, Tetrahedron Lett., 1987, 28, 6601–6604 CrossRef CAS; (c) A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411–2502 CrossRef CAS; (d) C. X. A. Da Silva, V. L. C. Gonçalves and C. J. A. Mota, Green Chem., 2009, 11, 38–41 RSC.
- (a) T. Cablewsky, A. F. Faux and C. R. J. Strauss, J. Org. Chem., 1994, 59, 3408 CrossRef CAS; (b) P. Sahai and R. A. J. Vishwakarma, J. Chem. Soc., Perkin Trans. 1, 1997, 12, 1845 Search PubMed; (c) J. S. Clarkson, A. J. Walker and M. A. Wood, Org. Process Res. Dev., 2001, 5, 630–635 Search PubMed; (d) B. Delfort, I. Durand, A. Jaecker, T. Lacome, X. Montagne, and F. Paille, US Patent, 0 163 949, 2003.
- (a) Alpharx INC. Pat., US2005/255 133 A1, 2005; (b) Ger. Offen 3447783, 198; (c) PCT Int. Appl. 88 050 71, 1988; (d) Etex Corporation, LEE, M. Youngmi, Patent, WO2005/117 919 A2, 2005; (e) K. Fraatz, and D. Mertin, Heep, Irish Patent, US2006/349 26 A1, 2006; (f) Celgene Corporation Patent, US2006/127 421 A1, 2006.
- (a) A. Jaecker-Voirol, I. Durand, G. Hillion, B. Delfort and X. Montagne, Oil Gas Sci. Technol., 2008, 63, 395–404 Search PubMed; (b) E. García, M. Laca, E. Pérez, A. Garrido and J. Peinado, Energy Fuels, 2008, 22, 4274–4280 CrossRef CAS.
- (a) J. L. Gras and J. F. Bonfanti, Synlett, 2000, 2, 248–250 Search PubMed; (b) B. E. Coleman, V. Cwynar, D. J. Hart, F. Havas, J. M. Mohan, S. Patterson, S. Ridenour, M. Schmidt, E. Smith and A. J. Wells, Synlett, 2004, 8, 1339–1342 Search PubMed.
- (a) D. L. Rachmankulov, S. S. Zlotskij, L. S. Rol'nik, G. T. Teregulova and H. J. Timpe, J. Prakt. Chem., 1989, 331, 697–699 Search PubMed; (b) R. Chenevert and R. Gagnon, J. Org. Chem., 1993, 58, 1054–1057 Search PubMed; (c) U. Naeser, A. J. Pierik, R. Scott, I. Cinkaya, W. Buckel and B. T. Golding, Bioorg. Chem., 2005, 33, 53–66 Search PubMed; (d) H. Zhang, K. Shibuya, H. Hemmi, T. Nishino and G. D. Prestwich, Org. Lett., 2006, 8, 943–946 Search PubMed; (e) W. Wu Li, M. Rongti, S. Subbalakshmi, H. J. Warshakoon, M. R. Kimbrell, M. W. Amolins, R. Ukani, A. Datta and S. A. David, J. Med. Chem., 2010, 53, 3198–3213 Search PubMed.
- (a) M. Selva and P. Tundo, Acc. Chem. Res., 2002, 35, 706–716 CrossRef CAS; (b) M. Selva, Pure Appl. Chem., 2007, 79, 1855–1867 CrossRef CAS.
- (a) M. Selva, C. A. Marques and P. Tundo, J. Chem. Soc., Perkin Trans. 1, 1994, 10, 1323–1328 Search PubMed; (b) A. Bomben, M. Selva, P. Tundo and L. Valli, Ind. Eng. Chem. Res., 1999, 38, 2075–2079 CrossRef CAS; (c) M. Selva, Synthesis, 2003, 18, 2872–2876 CrossRef; (d) M. Selva, P. Tundo and T. Foccardi, J. Org. Chem., 2005, 70, 2476–2485 CrossRef CAS; (e) M. Selva and P. Tundo, J. Org. Chem., 2006, 71, 1464–1470 CrossRef CAS; (f) M. Selva, P. Tundo, A. Perosa and D. Brunelli, Green Chem., 2007, 9, 463–468 RSC; (g) M. Selva, E. Militello and M. Fabris, Green Chem., 2008, 9, 73–79 Search PubMed; (h) M. Fabris, V. Lucchini, M. Noè, A. Perosa and M. Selva, Chem.–Eur. J., 2009, 15, 12273–12282 CrossRef CAS.
- (a) P. Tundo, F. Trotta and G. Moraglio, Ind. Eng. Chem. Res., 1988, 27, 1565–1571 CrossRef CAS; (b) B. Veldurthy, J.-M. Clacens and F. Figueras, J. Catal., 2005, 229, 237–242 CrossRef CAS.
- The structure of C5 and C6 isomers of both 2a–2b and 3a–3b were assigned through their MS spectra, and their amounts were evaluated by GC analysis (further details are in the ESI†).
- See references 10a, 11a, and also: M. Selva and A. Perosa, Green Chem., 2008, 10, 457–464 Search PubMed.
- (a) J. Otero and J. Nishikido, in Esterification Methods, Reactions, and Applications, Wiley-VCH, 2010 Search PubMed; (b) S. A. Pasias, N. K. Barakos and N. G. Papayannakos, Ind. Eng. Chem. Res., 2009, 48, 4266–4273 CrossRef CAS; (c) M. Diasakou, A. Louloudi and N. Papayannakos, Fuel, 1998, 77, 1297–1302 CrossRef CAS; (d) Z. Ilham and S. Saka, Lipid Technology, 2011, 23(1), 10–13 Search PubMed.
- (a) J. Radell and R. E. Rondeau, J. Chem. Eng. Data, 1971, 16, 104–105 Search PubMed; (b) J. Mercier, US Pat. 3, 928, 459 (Dec. 23, 1975); (c) J. March, in Advanced Organic Chemistry, 4th Ed. Wiley, 1992, p. 890 Search PubMed.
- The alcohol (methanol) derived from the DMC mediated alkylation reaction (Scheme 2). On the other hand, acidity for catalysis could be provided by the hygroscopic features of faujasites. See: Organic Solid State Reactions, ed. F. Toda, Kluwer Academic Publishers, Dordrecht, The Netherlands, 2002, pp. 160–161. Or to the presence of silanol groups on the surface of such zeolites Search PubMed; R. Ferwerda, J. H. van der Maas and P. J. Hendra, J. Phys. Chem., 1993, 97, 7331–7336 Search PubMed.
- J. T. D. Cross, R. Hunter and V. R. Stimson, Aust. J. Chem., 1976, 29(7), 1477–1481 Search PubMed.
- (a) M. Selva, M. Fabris and A. Perosa, Green Chem., 2011, 13, 863–872 RSC; (b) Y. Fu, H. Zhu and J. Shen, Thermochim. Acta, 2005, 434, 88–92 Search PubMed.
- Methanol could be ascribed to the hydrolysis of DMC due to traces of water present in the reaction mixture or adsorbed by the highly hygroscopic K2CO3. See J. S. Moya, E. Criado and S. De Aza, J. Mater. Sci., 1982, 17, 2213–2217 Search PubMed.
- P. Tundo, F. Aricò, A. E. Rosamilia and S. Memoli, Green Chem., 2008, 10, 1182–1189 RSC.
- Glyme had an excellent thermal stability and possessed physical properties comparable to DMC. See, MSDS for glyme on http://www.sigmaaldrich.com/catalog.
- With respect to GlyF, the use of solketal (available as a single 5-membered ring compound) appeared convenient to simplify both the analysis of the reaction mixtures with dialkyl carbonates and the characterization of the corresponding products.
- Both dibenzyl ether and benzyl alcohol did not derive from the reaction of solketal with DBnC. For, this reason, they were not indicated in Table 3.
- J. March, in Advanced Organic Chemistry, Reactions, Mechanisms and Structure, 4th Ed. Wiley and sons, 1992 Search PubMed.
- The extent of such side-reactions is not measurable under the conditions for the methylation of GlyF. In fact, a large excess of DMC is used and it also partially decomposes by a decarboxylation process (Scheme 5).
- G. Aknes, P. Albriktsen and P. Juvvik, Acta Chem. Scand., 1965, 19, 920–930 Search PubMed.
- P. Tundo, M. Selva, A. Perosa and S. Memoli, J. Org. Chem., 2002, 67, 1071–1077 CrossRef CAS.
- A. Y. Platonov, A. N. Evdokimov, A. V. Kurzin and H. D. Maiyorova, J. Chem. Eng. Data, 2002, 47, 1175–1176 Search PubMed Data allow only a rough estimation: the solubility of K2CO3 is reported at 180 °C for DMC, and at 25 °C for methanol, respectively. Since the solubility is expected to increase with the temperature, the comparison between DMC and MeOH would be even more in favor of the latter.
- (a) R. A. Van Santen and J. W. Niemantsverdriet, in Chemical Kinetics and Catalysis, Plenum Press, New York, 1995 Search PubMed; (b) G. C. Bond, in Heterogeneous Catalysis Principles and Applications, Oxford University Press, NewYork, USA, 2nd ed., 1987 Search PubMed.
- G. Maurin, P. L. Llewellyn and R. G. Bell, J. Phys. Chem. B, 2005, 109, 16084–16091 CrossRef CAS.
- H. Hayashi, J. Taniuchi, N. Furuyashiki, S. Sugiyama, S. Hirano, N. Shigemoto and T. Nonaka, Ind. Eng. Chem. Res., 1998, 37, 185–191 CrossRef CAS.
- The certified water content in DMC is ≤ 0.002% (from Aldrich). GlyF however, is defined as an hygroscopic liquid: see MSDS on http://www.sigmaaldrich.com/.
- F. Bonino, A. Damin, S. Bordiga, M. Selva, P. Tundo and A. Zecchina, Angew. Chem., Int. Ed., 2005, 44, 4774–4777 CrossRef CAS.
- (a) M. Selva, C. A. Alberto and P. Tundo, J. Chem. Soc., Perkin Trans. 1, 1995, 1, 1889–1893 Search PubMed; (b) A. Loris, A. Perosa, M. Selva and P. Tundo, J. Org. Chem., 2004, 69, 3953–3956 CrossRef CAS.
- (a) M. Selva, A. Perosa, F. Zordan and P. Tundo, Synlett., 2000, 1, 272–274; (b) M. Selva, A. Perosa and M. Fabris, Green Chem., 2008, 10, 1068–1077 RSC.
- J. F. King, G. T. Y- Tsang, M. M Abdel-Malik and N. C. Payne, J. Am. Chem. Soc., 1985, 107, 3224–3232 Search PubMed.
- D. D. Perrin and W. L. F. Armarego, in Purification of Laboratory Chemicals, 3rd Ed. Pergamon Press 1988 Search PubMed.
- MS spectra of both CO2 and DME gave a match quality higher than 97% when compared to standard products of the Wiley-MS database: Wiley Library of Mass Spectral Data, 2002, software of the GC-MS instrument.
- (a) H. Hill, A. Hill and H. Hibbert, J. Am. Chem. Soc., 1928, 50, 2242–2249 Search PubMed; (b) G. T. Teregulova, L. Z. Rol'nik, S. S. Zlot-skii, D. L. Rakhmankulov and N. N. Silishchev et al., J. Appl. Chem. USSR (Engl. Transl.), 1989, 62, 1620–1624 Search PubMed.
- (a) R. Chenevert and R. Gagnon, J. Org. Chem., 1993, 58, 1054–1057 Search PubMed; (b) V. Waagen, V. Partali, I. Hollingsaeter, M. S. S. Huang and T. Anthonsen, Acta Chem. Scand., 1994, 48, 506–510 Search PubMed; (c) Fujifilm Corp. Patent, EP1 795 208 A1, 2007; (d) G. Hirth and R. Barner, Helv. Chim. Acta, 1982, 65, 1059–1084 CAS; (e) M. W. Chun, D. H. Shin, H. R. Moon, J. Lee, H. Park and L. S. Jeong, Bioorg. Med. Chem. Lett., 1997, 7, 1475–1480 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis of dibenzyl carbonate (DBnC); GC calibration curves; MS spectra of componds 6c, 7c, 2a+2b and 3a+3b. See DOI: 10.1039/c1gc15796e |
| This journal is © The Royal Society of Chemistry 2012 |