Effective synthesis of 2,5-disubstituted tetrahydrofurans from glycerol by catalytic alkylation of ketones†
Magnus
Rueping
* and
Vilas B.
Phapale
RWTH Aachen University, Institute of Organic Chemistry, Landoltweg 1, 52074, Aachen, Germany. E-mail: Magnus.Rueping@RWTH-Aachen.de; Fax: (+49)-(0)241-8092665; Tel: 49 241 8094710
First published on 18th October 2011
Abstract
The [IrCl(cod)]2 catalyzed α-alkylation of substituted acetophenones with solketal followed by reduction and iron mediated cyclization provides 2,5-disubstituted tetrahydrofurans.
The development of improved protocols and strategies for the efficient construction of carbon-carbon bonds continues to be a challenge in organic synthesis. Among the vast array of established transformations, the α-alkylation of carbonyl compounds emerged as a powerful method for C–C bond formation.1 Typically classical methods make use of electrophilic alkylating agents like alkyl halides and result in the formation of undesired salts as by-products.2 As a greener and more economical alternative, the metal catalyzed α-alkylation of carbonyl, and its related compounds, with primary alcohols is attracting attention.3 This method possesses significant advantages over conventional α-alkylation methods as it proceeds through a hydrogen auto-transfer process (“borrowing hydrogen”) in which water is the only generated waste product.3 The overall process is a domino reaction based on the alcohol dehydrogenation which yields an aldehyde that undergoes aldol condensation with the existing carbonyl compound – commonly a methyl ketone – to afford an α,β-unsaturated carbonyl which is subsequently regioselectively reduced at the C–C double bond. Full reduction may also take place, affording a saturated secondary alcohol as the product. In this context, Cho et al. described the ruthenium catalyzed α-alkylation of ketones with primary alcohols leading to saturated secondary alcohols as major products.4 In order to suppress the reduction of the carbonyl group, the same reaction was performed in the presence of 1-dodecene serving as a hydrogen acceptor.5 Yus et al. reported the α-alkylation of ketones with benzylic alcohols using [Ru(DMSO)4Cl2] as a phosphine free catalyst.6 Heterogeneous palladium catalysts, such as palladium on carbon,7 and palladium nanoparticles on a polymer8 or entrapped in aluminium hydroxide [Pd/AlO(OH)]9 have also been applied in these processes.10
A catalytic system based on a [Ir(cod)Cl]2–PPh3–KOH combination was reported to be effective for the alkylation of ketones with primary alcohols and diols in the absence of solvent.11Iridium was also found to be an efficient catalyst for the α-alkylation of other functional groups such as nitriles or acetates.12–14
Given the features of the described transformations, we decided to investigate the possibility of integrating such a process into a more elaborate sequential protocol which would allow the synthesis of more complex structures in a sustainable manner. Herein, we report our designed strategy and its successful application in the diastereoselective synthesis of 2,5-disubstituted tetrahydrofurans (Scheme 1).15
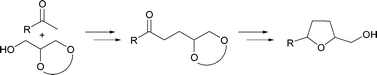 | ||
| Scheme 1 Proposed sequential strategy for the synthesis of tetrahydrofuran derivatives. | ||
Due to its latent functionalization, solketal (2), which is readily available through the protection of glycerol with acetone,16 was chosen as a hydrogen donor for the α-alkylation of acetophenone. A large range of bases was evaluated in the reaction of acetophenone (1a) with solketal (2) and the results are summarized in Table 1.
|
|
||
|---|---|---|
| Entry | Base | Yield (%)b |
| a 1a (1 mmol) was reacted with solketal (2, 1.8 mmol) in the presence of Ir complex (2 mol%) and base (10 mol%) in toluene (0.2 mL) at 110 °C for 17 h. b Yield – numbers in the brackets represent the mixture of α-alkylated ketone (major) and alcohol. c LiOH·H2O (40 mol%) was used. | ||
| 1 | K2CO3 | 8 |
| 2 | CsOH | 25 |
| 3 | NaOH | 48 (36 + 12) |
| 4 | Ba(OH)2 | 53 (46 + 7) |
| 5 | KOH | 61 (55 + 6) |
| 6 | LiOH·H2O | 68 (60 + 8) |
| 7c | LiOH·H2O | 82 (73 + 9) |
| 8 | Et3N | 0 |
| 9 | — | 0 |
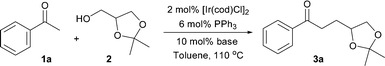
In the presence of K2CO3, the reaction of acetophenone with solketal proceeded with less than 10% conversion (Table 1, entry 1). Stronger bases like CsOH, NaOH and Ba(OH)2 afforded the product in moderate yields (Table 1, entries 2–4). LiOH·H2O (40 mol%) proved to be the most successful base affording the desired product in 82% yield (Table 1, entry 7). In the presence of triethylamine no alkylation product could be detected (Table 1, entry 8). The same result was observed when the reaction was performed in the absence of a base.
With the optimized conditions for the alkylation reaction in hand, the reaction of various acetophenones with solketal was investigated using catalytic amounts of [Ir(cod)Cl]2 (Table 2). Subsequent reduction with NaBH4 afforded the corresponding secondary alcohols 4 in good to high yields. Acetal deprotection and cyclization mediated by FeCl3 afforded 2,5-disubstituted tetrahydrofurans 5 bearing a primary alcohol functional group in very good yields.17
|
|
|||
|---|---|---|---|
| Entry | 1, Ar | 4, Yield (%)a,b |
5, Yield (%)c (trans![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :cis)d,e :cis)d,e |
| a The reactions were carried out with 1 (1 mmol), solketal (2, 1.8 mmol) in the presence of [Ir(cod)Cl]2 (2 mol%), PPh3 (6 mol%) and LiOH·H2O (40 mol%) in toluene (0.2 mL) at 110 °C; solvent was removed and 3 was treated with 1 equiv. of NaBH4 in methanol at 10 °C for 2 h. b Yield over two steps. c Alcohol 4 was treated with 1 equiv. of anhydrous FeCl3 in DCM at room temperature for 45 min. d Diastereomeric ratio was determined by 1H NMR spectroscopy. e The trans stereochemistry for the major diastereomer has been assigned according to the literature.17e,f,h | |||
| 1 | Ph | 80 |
5a, 86 (1.8:1) |
| 2 | 4-F–C6H4 | 87 |
5b, 88 (2.0:1) |
| 3 | 3-CF3–C6H4 | 82 |
5c, 81 (1.4:1) |
| 4 | 4-C6H5–C6H4 | 93 |
5d, 89 (6.3:1) |
| 5 | 4-t-Bu–C6H4 | 85 |
5e, 91 (3.8:1) |
| 6 | 3,4-CH3–C6H3 | 88 |
5f, 92 (3.3:1) |
| 7 | 4-MeO–C6H4 | 86 |
5g, 90 (1.8:1) |
| 8 |
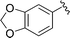
|
91 |
5h, 89 (1.7:1) |
| 9 |
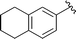
|
86 |
5i, 88 (4.3:1) |
| 10 |
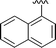
|
84 |
5j, 83 (5.9:1) |
| 11 |
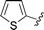
|
77 |
5k, 74 (1:1) |
Moderate trans/cis isomeric ratios were observed with ketones bearing electron-withdrawing (Table 2, entries 2, 3) and electron-donating groups (Table 2, entries 7, 8) on the aryl substituent. Slightly better trans/cis selectivities were observed with substrates bearing t-Bu and Me groups (Table 2, entries 5, 6). Improved diastereomeric ratios were obtained for the substrates bearing biphenyl and naphthyl residues. (Table 2, entries 4, 10). The heteroaryl derivative 1k afforded the desired product 5k as a 1:1 diastereomeric mixture in good yields (Table 2, entry 11). Cyclic ketone 1l worked well in the alkylation and reduction steps (4l 73% yield), however, the yield of the final step was moderate and the product was obtained as a 1:1 ratio of isomers (Scheme 2, 5l 46% yield).
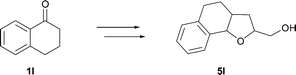 | ||
| Scheme 2 Conversion of cyclic ketone 1l. | ||
Furthermore, a one-pot three-step procedure that does not require chromatographic purification of any of the intermediates, proved to be feasible and the product 5a has been obtained in an overall yield of 59%.
Subsequent derivatisation of the products is possible through oxidation of the primary alcohol to afford 5-phenyltetrahydrofuran-2-carbaldehydes which can easily be converted into the corresponding six-membered ring lactones 6 (Scheme 3).18
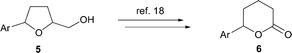 | ||
| Scheme 3 Functionalization of the tetrahydrofuran derivatives 5. | ||
Regarding the reaction mechanism, it is assumed that the alkylation reaction follows the same pathway as the previously reported related transformations (Scheme 4).3 In the first step, hydrogen transfer oxidation of the alcohol 2 gives aldehyde 7 and an iridium hydride. Base-catalyzed aldol condensation between aldehyde 7 and ketone 1 gives α,β-unsaturated ketone 8 which is hydrogenated by the Ir-hydride complex generated in the course of the reaction, to form the α-alkylated ketone 3.
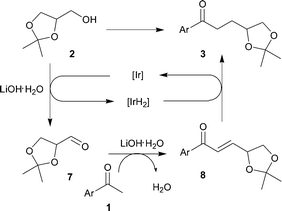 | ||
| Scheme 4 Proposed mechanism for the α-alkylation of solketal. | ||
In conclusion, a convenient and highly effective Ir-catalyzed α-alkylation of ketones with subsequent reduction and deprotection/cyclization methodology for the synthesis of valuable 2,5-disubstituted tetrahydrofurans was developed. Notably, commercially available substituted acetophenones as well as solketal a readily available derivative of the bulk chemical glycerol are used in the atom-economic transfer hydrogen reaction, whereby water is the only by-product. The subsequent reduction and deprotection/cyclization proceed under mild reaction conditions and afford 2,5-disubstituted tetrahydrofurans in good to high overall yields. In addition, a more practical, one-pot three-step procedure has been developed.
Acknowledgements
V.B.P. thanks the Alexander von Humboldt foundation for a post-doctoral research fellowship.Notes and references
- (a) D. Caine, in Comprehensive Organic Synthesis, vol. 3, ed. B. M. Trost and I. Fleming, Pergamon, Oxford, 1991, pp. 1–63 Search PubMed; (b) S. Carrettin, J. Guzman and A. Corma, Angew. Chem., Int. Ed., 2005, 44, 2242–2245 CrossRef CAS; (c) Modern Carbonyl Chemistry, ed. J. Otera, Wiley-VCH, Weinheim, 2000 Search PubMed.
- Handbook of Green Chemistry and Technology, ed. J. H. Clark and D. J. Macquarrie, Blackwell, Oxford, 2002 Search PubMed.
- For recent reviews (a) G. Guillena, D. J. Ramón and M. Yus, Angew. Chem., Int. Ed., 2007, 46, 2358–2364 CrossRef CAS; (b) T. D. Nixon, M. K. Whittlesey and J. M. J. Williams, Dalton Trans., 2009, 753–762 RSC; (c) M. H. S. A. Hamid, P. A. Slatford and J. M. J. Williams, Adv. Synth. Catal., 2007, 349, 1555–1575 CrossRef CAS; (d) F. Alonso, F. Foubelo, J. C. Gonzáles-Gómez, R Martínez, D. J. Ramón, P. Riente and M. Yus, Mol. Diversity, 2010, 14, 411–424 Search PubMed.
- (a) C. S. Cho, B. T. Kim, T.-J. Kim and S. C. Shim, J. Org. Chem., 2001, 66, 9020–9022 CrossRef CAS. For a combined Ir–Ru catalytic system applied in the asymmetric alkylation of ketones with alcohols affording chiral secondary alcohols in good yields and excellent selectivities, see: (b) G. Onodera, Y. Nishibayashi and S. Uemura, Angew. Chem., Int. Ed., 2006, 45, 3819–3822 CrossRef CAS.
- C. S. Cho, B. T. Kim, T.-J. Kim and S. C. Shim, Tetrahedron Lett., 2002, 43, 7987–7989 CrossRef CAS.
- (a) R. Martínez, G. J. Brand, D. J. Ramón and M. Yus, Tetrahedron Lett., 2005, 46, 3683–3686 CrossRef CAS; (b) R. Martínez, D. J. Ramón and M. Yus, Tetrahedron, 2006, 62, 8988–9001 CrossRef CAS.
- C. S. Cho, J. Mol. Catal. A: Chem., 2005, 240, 55–60 CAS.
- Y. M. A. Yamada and Y. Uozumi, Org. Lett., 2006, 8, 1375–1378 CrossRef CAS.
- M. S. Kwon, N. Kim, S. H. Seo, I. S. Park, R. K. Cheedrala and J. Park, Angew. Chem., Int. Ed., 2005, 44, 6913–6915 CrossRef CAS.
- For the alkylation of methyl ketones with primary alcohols promoted by Nickel nanoparticles (1 equiv), see: F. Alonso, P. Riente and M. Yus, Synlett, 2007, 1877–1880 Search PubMed.
- (a) K. Taguchi, H. Nakagawa, T. Hirabayashi, S. Sakaguchi and Y. Ishii, J. Am. Chem. Soc., 2004, 126, 72–73 CrossRef CAS . For the reaction of ketones with diols leading to diketones, see; (b) K. Maeda, Y. Obora, S. Sakaguchi and Y. Ishii, Bull. Chem. Soc. Jpn., 2008, 81, 689–696 Search PubMed.
- Iridium catalyzed alkylation of nitriles: (a) C. Löfberg, R. Grigg, M. A. Whittaker, A. Keep and A. Derrick, J. Org. Chem., 2006, 71, 8023–8027 CrossRef . Rhodium catalyzed alkylation of nitriles; (b) R. Grigg, T. R. B. Mitchell, S. Sutthivaiyakit and N. Tongpenyai, Tetrahedron Lett., 1981, 22, 4107–4110 CrossRef CAS . Ruthenium catalyzed alkylation of nitriles; (c) K. Motokura, D. Nishimura, K. Mori, T. Mizugaki, K. Ebitani and K. Kaneda, J. Am. Chem. Soc., 2004, 126, 5662–5663 CrossRef CAS.
- Iridium catalyzed alkylation of acetates: Y. Iuchi, Y. Obora and Y. Ishii, J. Am. Chem. Soc., 2010, 132, 2536–2537 Search PubMed.
- For the Ir catalyzed alkylation of other substrates with alcohols (C–C bond formation): (a) M. G. Edwards and J. M. J. Williams, Angew. Chem., Int. Ed., 2002, 41, 4740–4743 CrossRef CAS; (b) C. Löfberg, R. Grigg, A. Keep, A. Derrick, V. Sridharan and C. Kilner, Chem. Commun., 2006, 5000–5002 RSC; (c) P. J. Black, G. Cami-Kobeci, M. G. Edwards, P. A. Slatford, M. K. Whittlesey and J. M. J. Williams, Org. Biomol. Chem., 2006, 4, 116–125 RSC; (d) M. Morita, Y. Obora and Y. Ishii, Chem. Commun., 2007, 2850–2852 RSC; (e) S. Whitney, R. Grigg, A. Derrick and A. Kepp, Org. Lett., 2007, 9, 3299–3302 CrossRef CAS; (f) K.-I. Fujita, C. Asai, T. Yamaguchi, F. Hanasaka and R. Yamaguchi, Org. Lett., 2005, 7, 4017–4019 CrossRef CAS; (g) D. Gnanamgari, C. H. Leung, N. D. Schley, S. T. Hilton and R. H. Crabtree, Org. Biomol. Chem., 2008, 6, 4442–4445 RSC.
- For a recent review on the diastereoselective synthesis of 2,5-disubstituted tetrahydrofuranes, see: G. Jalce, X. Franck and B. Figadère, Tetrahedron: Asymmetry, 2009, 20, 2537–2581 Search PubMed.
- (a) C. X. A. da Silva, V. L. C. Goncalves and C. J. A. Mota, Green Chem., 2009, 11, 38–41 RSC; (b) A. Behr, J. Eilting, K. Irawadi, J. Leschinski and F. Lindner, Green Chem., 2008, 10, 13–30 RSC.
- For alternative protocols, see: (a) S. Inoki and T. Mukaiyama, Chem. Lett., 1990, 67–70 CrossRef CAS; (b) J. Berninger, U. Koert, C. Eisenberg-Höhl and P. Knochel, Chem. Ber., 1995, 128, 1021–1028 Search PubMed; (c) K. Miura, T. Hondo, S. Okajima and A. Hosomi, Tetrahedron Lett., 1996, 37, 487–490 CrossRef CAS; (d) K. Miura, T. Hondo, T. Takahashi and A. Hosomi, Tetrahedron Lett., 2000, 41, 2129–2132 CrossRef CAS; (e) K. Miura, S. Okajima, T. Hondo, T. Nakagawa, T. Takahashi and A. Hosomi, J. Am. Chem. Soc., 2000, 122, 11348–11357 CrossRef CAS; (f) J. Hartung, S. Drees, M. Greb, P. Schmidt, I. Svoboda, H. Fuess, A. Murso and D. Stalke, Eur. J. Org. Chem., 2003, 2388–2408 CrossRef CAS; (g) B. M. Pérez, D. Schuch and J. Hartung, Org. Biomol. Chem., 2008, 6, 3532–3541 RSC; (h) C. Palmer, N. A. Morra, A. C. Stevens, B. Bajtos, B. P. Machin and B. L. Pagenkopf, Org. Lett., 2009, 11, 5614–5617 Search PubMed.
- L. Wang, K. Thai and M. Gravel, Org. Lett., 2009, 11, 891–893 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: General procedures, characterization data, and copies of the NMR spectra of the products are provided. See DOI: 10.1039/c1gc15764g |
| This journal is © The Royal Society of Chemistry 2012 |