5-Membered N-heterocyclic compounds by dimethyl carbonate chemistry†
Fabio
Aricò
,
Umberto
Toniolo
and
Pietro
Tundo
*
Università Ca' Foscari di Venezia, Dipartimento Scienze Ambientali, Informatica e Statistica, Dorsoduro 2137, 30123, Venice, Italy. E-mail: tundop@unive.it; Fax: + 39 041234 8620; Tel: +39 041234 864
First published on 18th October 2011
Abstract
Aliphatic and aromatic 1,4-bifunctional compounds bearing a primary alcoholic function and an amine can be efficiently cyclised with dimethyl carbonates in the presence of a base to achieve 5-membered N-heterocyclic compounds. This novel synthetic pathway is quantitative, one-pot and green as it does not involve the use of chlorine solvents or reagent.
N-Based heterocycles are very abundant in nature since they are present as structural subunits in many natural products such as vitamins, hormones and alkaloids.1 These compounds are also interesting from an industrial point of view especially for the synthesis of pharmaceuticals, herbicides, pesticides, dyes, etc.1
Among the reaction pathways leading to nitrogen-containing heterocycles, many involve heavy metals, e.g. metal-catalyzed intramolecularcyclisation of aliphatic α,ω-diamine,2intramolecular and intermolecularRu-catalyzed reactions,3intramolecularcyclisation of diallyl amine by Grubb's catalyst,4 or gas-phase high-temperature reaction using zeolites.5 In recent years, more sustainable approaches for the synthesis of N-based heterocycles have been reported,6 such as photocatalytic cyclisation of α,ω-diamine carboxylic acids by aqueous semiconductor suspensions7 and microwave-assisted synthesis from alkyl dihalides and primary amines.8
However, most of the above-mentioned reactions still require high temperature and long reaction time, utilize chlorine-based chemistry or eventually organic chlorinated solvents.
Short chain dialkyl carbonates such as dimethyl carbonate (DMC), produced nowadays by clean processes,10 are renowned for possessing properties of low toxicity and high biodegradability, which make them true green solvents and reagents.11 DMC has been used as efficient eco-sustainable substitute of the most common methylating and carboxymethylating agents such as phosgene, methyl halides or methylsulfate that are toxic and highly corrosive.12 Dialkyl carbonates and in particular DMC have shown high selectivity with different monodentate and bidentate nucleophiles acting as methylating and/or carboxymethylating agent.13 The reactivity of the two electrophilic centers of DMC can be selectively tuned, temperature being the key factor. In fact, usually at reflux temperature (T = 90 °C) DMC acts as methoxycarbonylation agent by BAc2 mechanism while at higher temperature (T > 150 °C) the methylation reaction occurs via the BAl2 mechanism. Both reactions produce as by-product only methanol and eventually CO2.11–13
Exploiting the DMC (BAl2-BAc2) chemistry, recently we reported a novel, one-pot, environmentally benign and chlorine-free synthetic pathways for the synthesis of 5-membered cyclic ethers by intramolecularcyclisation of 1,4-diols.9
In this work, we report a DMC-promoted intramolecularcyclisation for the selective synthesis of 5-membered N-based cyclic molecules.
4-Amino-1-butanol 1 was selected as starting material as it is the simplest aliphatic model available for this study. It is noteworthy thatthe family of the carboxyalkyl pyrrolidine has been recently used as key intermediates in the synthesis of heterocyclic arylsulphones that showed to be efficient in the treatment of diseases of the central nervous system, e.g. Alzheimer's disease and schizophrenia.4
Preliminary experiments on this substrate were carried out using DMC as solvent and reagent with different catalysts (Table 1): metallic homogenous catalysts (entries 1–2), alkali carbonates (entries 3–4), heavy metal basic carbonates (entry 5), strong base (entry 6) and hydrotalcite14 (entry 7).‡
| Entry | Catalyst | Time (h) | Conversion (%) | N-Methoxycarbonyl pyrrolidine b (%) |
|---|---|---|---|---|
| a All reactions were carried out in autoclave. b Yields were calculated by GC-MS in the presence of an internal standard (decane); in all the cases the methyl 4-(methoxycarbonyloxy)butylcarbamate (its anion 3 is shown in Scheme 1) was the only other product observed. | ||||
| 1 | Zn(OAc)2 | 3 | 100 | 34.7 |
| 2 | SnOBu2 | 3 | 100 | 27.5 |
| 3 | K2CO3 | 3 | 100 | 49.3 |
| 4 | Cs2CO3 | 3 | 100 | 62.3 |
| 5 | (ZnCO3)2·[Zn(OH)2]3 | 3 | 100 | 38.7 |
| 6 | MeONa | 3 | 100 | 46.0 |
| 7 | HT KW2000 | 3 | 100 | 47.8 |
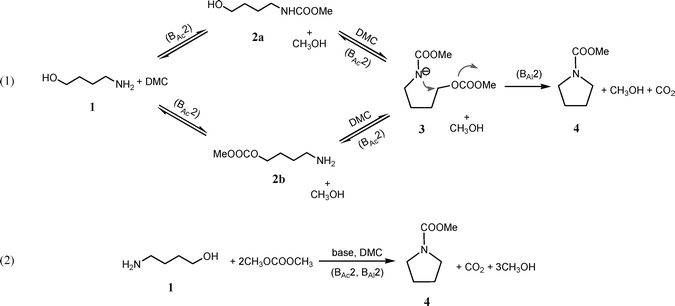 | ||
| Scheme 1 Intramolecular N-heterocyclisation of 4-amino-1-butanol 1 to form the N-carboxymethyl pyrrolidine 4; (eqn (1)) Reaction mechanism; (eqn (2)) Stoichiometry of the reaction. | ||
All the reactions were conducted in autoclave with catalytic amount of base/catalyst and in the presence of DMC.
Results collected showed in any case the formation of the N-methoxycarbonyl pyrrolidine 4 from modest to good yields (34–62%).
Among the catalysts used, alkali carbonates and in particular Cs2CO3 resulted the more efficient ones (62% yield). In any case the only by-product formed was the methyl 4-(methoxycarbonyloxy)butylcarbamate that was isolated as pure and fully characterised.‡
This reaction is a remarkable example of hard-soft acid–base theory applied to the DMC, as the starting substrate include two different nucleophiles, i.e. a primary amine and a primary alcohols, that discriminate between the two electrophilic centers of the DMC leading to the carboxymethyl pyrrolidine by one-pot cyclisation.
Most probably the reaction proceeds by a sequence of carboxymethylation and alkylation reactions (Scheme 1): the 4-amino-1-butanol 1 firstly undergoes carboxymethylation at the hydroxyl (or amine) group (BAc2), then, or most probably at the same time, the amine (or hydroxyl) moiety will also carboxymethylate (BAc2). As a consequence, the amino group of the so formed carbamate (its related anion 3 is shown in Scheme 1) results softer in character, as demonstrated by previous investigations,11c and it undergoes fast alkylation to form selectively the carboxymethyl pyrrolidine 4 (BAl2).15 It is noteworthy that the formation of the N-based cyclic 4 is favoured because all the reactions (BAc2) depicted in Scheme 1 are of equilibrium, except the one related to the cyclic formation (BAl2).
Further investigations of the 4-amino-1-butanol 1 were also conducted at reflux temperature and atmospheric pressure using a base and DMC as solvent and reagent. Results collected are reported in Table 2. High-yielding N-heterocyclisation of the starting material was achieved by employing potassium tert-butoxide as base (entries 1–2). Most probably the excess of base used in the reaction is needed for the three subsequent reactions to take place (Scheme 1). In fact, performing the reaction using only 0.5 eq mol of base at higher temperature also in autoclave resulted in lower yield (entry 3).
| Entry | Temp. (°C) | KOtBu (eq. mol) | Conv. (%) | N-Methoxycarbonyl pyrrolidine a (%) |
|---|---|---|---|---|
| a Yields were calculated by 1H NMR spectrometry; b Reaction conducted in autoclave. | ||||
| 1 | 90 | 2 | 100 | 77 |
| 2 | 90 | 2.5 | 100 | 86 |
| 3 | 160b | 0.5 | 100 | 76 |
Investigations were also conducted on two simple aromatic bifunctional nucleophile i.e.2-(2-aminophenyl)ethanol and 2-(aminomethyl)benzyl alcohol (Scheme 2). Results, reported in Table 3 and Table 4, respectively, demonstrated that also in this case N-carboxymethyl indoline 6 and N-carboxymethyl isoindoline 9 were formed in quantitative yield by intramolecularcyclisation.
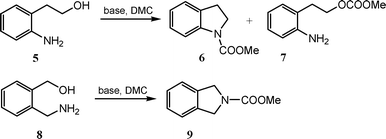 | ||
| Scheme 2 Synthesis of N-carboxymethyl indoline 6 and N-carboxymethyl isoindoline 9. | ||
| Entry | Base (eq. mol) | Temp. (°C) | Conv. (%) | Carboxymethyl indoline (GC-MS%) |
|---|---|---|---|---|
| a The reaction time is 6 hours, yields were calculated by GC-MS data, the intermediate 6 was the only other compounds observed. b Using DMC recycled from entry 1 (Table 3). c Isolated yield. d In autoclave. | ||||
| 1 | NaOMe (2.5) | 90 | 100 | 82 |
| 2b | NaOMe (2.5) | 90 | 100 | 83 |
| 3 | KOtBu (2.5) | 90 | 100 | 95 (78)c |
| 4d | KOtBu (0.1) | 180 | 100 | 93 |
In particular, when 2-(2-aminophenyl)ethanol 5 was used as substrate the products formed were the carboxymethyl indoline 6 and small amount of the cyclisation intermediate 2-aminophenethyl methylcarbonate 7, the only intermediate observed (entries 1, 3 Table 3).‡Column chromatography of the reaction mixture allowed isolation of the pure compounds and their characterisation. It is noteworthy that in all the above-mentioned reactions DMC is employed in excess since it serves as solvent and reagent, however it can be easily recycled after filtration of the reaction mixture and evaporation under vacuum. In fact, performing the cyclisation reaction by using recycled DMC (entry 2, Table 3) resulted in the high yielding conversion of the starting material into the carboxymethyl indoline 5.‡ Furthermore, when the cyclisation reaction was conducted in autoclave at high temperature and in the presence of catalytic amount of base (entry 4 Table 3), carboxymethyl indoline formed, once again, in good yield (83%).
Table 4 reports the results achieved for the cyclisation of 2-(aminomethyl)benzyl alcohol 8. This substrate was synthesised by reduction of ethyl 2-cyanobenzoate according to literature procedure.16 When the cyclisation reaction of 8 was conducted in the presence of a strong base (2.5 eq. mol.) at reflux conditions, the carboxymethyl isoindoline 9 formed in quantitative yield as sole product. (entry 1–2, Table 4). It is also possible to carry out the reaction using catalytic amount of base (0.1 eq. mol.), but this require the use of autoclave and high temperature (entry 3, Table 4)
The reaction of aliphatic and aromatic 4-amino-1-butanol compounds with DMC in the presence of a base and in mild condition led to the corresponding N-based cyclic in high yield and short reaction time. The formation of the N-based cyclic 4, 6 and 9 is favoured due to the reaction mechanism comprising of several equilibrium reactions (BAc2), meanwhile the cyclic formation (BAl2) is the only kinetically driven reaction (Scheme 1). The cyclisation reaction was conducted at reflux condition in the presence of 2.5 eq. mol. of base or utilizing a catalytic amount of base (0.1 eq. mol.) in an autoclave. Both reactions resulted in the high yielding formation of the 5-membered N-heterocyclic compound, although using small amount of base required higher temperature.
Comparing this reaction with the other avaiable synthetic pathways, the DMC-mediated reaction is green, high yielding, occurs in one step, do not require any chlorine-based chemical or strong acid and do not produce any chlorinated waste material. DMC, employed as solvent and reagent in the cyclisation reaction, can be easily recovered by distillation and reused. General applicability of the new synthesis on other suitable substrates is currently under investigation.
References
- (a) A. R. Katritzky, C. A. Ramsden, J. A. Joule and V. V. Zhdankin, Handbook of Heterocyclic Chemistry, Elsevier, 2010 Search PubMed; (b) A. Padwa and S. Bur, Chem. Rev., 2004, 104, 2401 CrossRef CAS.
- (a) K. Kindler and D. Matthies, Chem. Ber., 1962, 95, 1992 Search PubMed; (b) T. M. Gadda, X.-Y. Yu and A. Miyazawa, Tetrahedron, 2010, 66, 1249 Search PubMed.
- (a) M. Haniti, S. A. Hamid, C. Liana Allen, G. W. Lamb, A. C. Maxwell, H. C. Maytum, A. J. Watson and J. M. J. Williams, J. Am. Chem. Soc., 2009, 131, 1766 CrossRef CAS; (b) S.-I. Murahashi, K. Kondo and T. Hakata, Tetrahedron Lett., 1982, 23, 229 CrossRef CAS.
- R. Grandel, W. M. Braje, A. Haupt, S. C. Turner, U. Lange, K. Dresher, L. Unger and D. Plata, patent WO 2007/118899 A1, publication date 25.10.2007 to ABBOTT GMBH & CO.KG.
- R. V. Radha, N. Srinivas, S. J. Kulkami and K. V. Ragavan, Ind. J. Chem., 1999, 38A, 286 Search PubMed.
- N. R. Candeias, L. C. Branco, P. M. P. Gois, C. A. M. Afonso and A. F. Trindade, Chem. Rev., 2009, 109, 2703 CrossRef CAS.
- B. Ohtani, S. Tsuru, S. Nishimoto and T. Kagiya, J. Org. Chem., 1990, 55, 5551 CrossRef CAS.
- Y. Ju and R. S. Varma, J. Org. Chem., 2006, 71, 135 CrossRef CAS.
- H. S. Bevinakatti, C. P. Newman, S. Elwood, P. Tundo and F. Arico, Cyclic Ethers. International Patent PCT/GB2008/050567, 22 January, 2009.
- (a) Asahi Kasei Chemicals Corporation Patent, WO2007/34669 A1, 2007; (b) The Merck Index, 11th edn, ed. S Budavari, Merck and Co. Inc., Ralway, NJ, 1989 Search PubMed; (c) M Wang, H Wang, N Zhao, W Wei and Y Sun, Ind. Eng. Chem. Res., 2007, 46, 2683 Search PubMed.
- (a) P. Tundo and M. Selva, Acc. Chem. Res., 2002, 35, 706 CrossRef CAS; (b) P. Tundo, M. Selva, A. Perosa and S. Memoli, J. Org. Chem., 2002, 67, 1071 CrossRef CAS; (c) A. E. Rosamilia, F. Aricò and P. Tundo, J. Org. Chem., 2008, 73(4), 1559 CrossRef CAS; (d) P. Tundo, S. Memoli, D. Hérault and K. Hill, Green Chem., 2004, 6, 609 RSC; (e) P. Tundo, F. Aricò, A. E. Rosamilia and S. Memoli, Green Chem., 2008, 10, 1182 RSC.
- L. Cotarca and H. Ecket, in Phosgenations – A Handbook, Wiley-VCH, Verlag GmbH & Co., 2003 Search PubMed.
- A. E. Rosamilia, F. Aricò and P. Tundo, J. Phys. Chem. B, 2008, 112, 14525 Search PubMed.
- F. Bonino, A. Damin, S. Bordiga, M. Selva, P. Tundo and A. Zecchina, Angew. Chem., Int. Ed., 2005, 44, 4774 CrossRef CAS.
- In principle the reaction could also leads to the formation of tetrahydrofuran (via elimination of NHCOOCH3 by BAl2 mechanism), however this product was never observed.
- R. Cao, P. Mueller, S. J. Lippard and Stephen, J. Am. Chem. Soc., 2010, 132(49), 17366 Search PubMed.
- A. Minatti and S. L. Buchwald, Org. Lett., 2008, 10, 2721 CrossRef CAS.
- D. Piet, S. de Bruijn, J. Sum and G. Lodder, J. Heterocycl. Chem., 2005, 42, 227 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectra, GC-MS analysis. See DOI: 10.1039/c1gc15698e |
| ‡ Synthesis of carboxymethyl pyrrolidine in autoclave (Table 1): in a typical experiment 4-amino 1-butanol (0.26 mL, 2.80 mmol), DMC (10 mL) and 10% mol of catalyst were heated at T = 180 °C while stirring continuously under nitrogen atmosphere for three hours. Results were collected by gas chromatography in the presence of an internal standard (decane). Gradient elution chromatography using Et2O/hexane (3/2) on silica gel allowed all of the products to be isolated as pure compounds. Carboxymethyl pyrrolidine 4: analysis conducted on the isolated product were consistent with the one present in the literature.3b4-(Methoxycarbonyloxy)butylcarbamate: as white oil C8H15NO5; M = 205.2084 g mol−1; 1H NMR (300 MHz, CD3CN) δ = 1.5–1.72 (m, 4H), 3.12 (7, 2H), 3.6 (s, 3H), 3.75 (s, 3H), 4.12 (t, 2H), 5.7 (s, 1H); 13C NMR (75 MHz, CD3CN) δ = 155.5, 67.3, 54.1, 51.1, 39.9, 25.8, 25.5. Synthesis of carboxymethyl pyrrolidine at reflux conditions (Table 2): in a typical experiment 4-amino 1-butanol (0.5 mL, 5.60 mmol), DMC (15 mL) and potassium tert-butoxide were heated at T = 90 °C while stirring continuously under nitrogen atmosphere for three hours. The reaction outcome was followed by 1H NMR spectrometry (see ESI).† Synthesis of carboxymethyl (iso)indoline (Table 3, Table 4): in a typical experiment the substrate, i.e.2-(2-amino phenyl)ethanol (0.5 mL, 3.64 mmol), DMC (15 mL) and potassium tert-butoxide (2.5 eq. mol.) were heated at T = 90 °C while stirring continuously under nitrogen atmosphere for six hours. The reaction outcome was followed by GC-MS analysis. If necessary, gradient elution chromatography using hexane/EtOAc (5/2) on silica gel allowed isolation of the pure carboxymethyl indoline 5 and of a small amount of the intermediate 2-aminophenethyl methylcarbonate 6. Carboxymethyl indoline: Analysis conducted on the isolated product were consistent with the one obtained present in the literature.17 Mp 69–72 °C (lit. mp 68–72 °C).17 2-Aminophenethyl methylcarbonate 7: as a light red oil C10H13NO3 M = 195.2 g mol−1; 1H NMR (400 MHz, CDCl3) δ = 2.88–2.92 (t, 2H), 3.76 (s, 3H), 4.28–4.32 (t, 2H), 6.71–6.74 (m, 2H), 7.1–7.26 (m, 2H); 13C NMR (100 MHz, CDCl3) δ = 155.9, 145.0, 130.4, 128.0, 121.1, 118.7, 115.9, 67.0, 54.8, 31.0. Carboxymethyl isoindoline: analyses conducted on the isolated product were consistent with the one obtained present in the literature.18 Synthesis of carboxymethyl indoline with recycled DMC (entry 2, Table 3): DMC was distilled from the reaction mixture (entry 1, Table 3). Pure DMC (5 mL) was added to the recovered DMC (10 mL) in order to have enough solvent/reagent to conduct the experiment (15 mL). The reaction was then conducted in the same conditions reported for the synthesis of carboxymethyl indoline 6. |
| This journal is © The Royal Society of Chemistry 2012 |