Improved direct production of 2,3-dimethylbutenes and 3,3-dimethylbutene from 2-methylpropene on tungsten hydride based catalysts†
Anthony
Garron
,
François
Stoffelbach‡
,
Nicolas
Merle
,
Kai C.
Szeto
,
Jean
Thivolle-Cazat
,
Jean-Marie
Basset§
,
Sébastien
Norsic
and
Mostafa
Taoufik
*
Université Lyon 1, Institut de Chimie de Lyon, CPE Lyon, CNRS, UMR 5265 C2P2, LCOMS, Bâtiment 308, F 43 Blvd du 11 Novembre 1918 F-69616, Villeurbanne Cedex, France. E-mail: taoufik@cpe.fr; Fax: +33 (0)4 72 43 17 95
First published on 23rd August 2012
Abstract
2-Methylpropene in the presence of W–H/Ni1%–Al2O3-(500) is transformed in high selectivity into a mixture of 2,3-dimethylbutenes (2,3-DMBs = DMB-1 and DMB-2) and neohexene. 2,3-DMBs arise from the unfavoured 2-methylpropene self-metathesis reaction whereas the neohexene originates from a cascade reaction: 2-methylpropene dimerisation followed by cross metathesis.
Branched C6 hydrocarbons are valuable products within broad domains (fine and petrochemical industry). In fact, 3,3-dimethyl-1-butene (neohexene) is used in the preparation of fine chemicals such as aromatic musks, which are widely used in perfumery, household soaps and detergents.1,2 For example, 1,1,3,4,4,6-hexamethyl-1,2,3,4-tetrahydronaphthalene (Tonalide®) is classically synthesized by acid-catalysed cycloalkylation via Friedel–Crafts acylation of p-cymene with neohexene.2 This is currently prepared on a large scale only by Phillips Petroleum from 2-methylpropene (isobutene) and proceeds in two steps and in separate reactors.3 The first reaction comprises the dimerisation of isobutene into a mixture of 2,4,4-trimethylpent-2-ene (DIB-2) and 2,4,4-trimethylpent-1-ene (DIB-1) on an acidic catalyst (eqn (1), Scheme 1). The production of neohexene is then performed on the dual catalyst WO3/SiO2 and MgO. WO3/SiO2 catalyses the cross metathesis of DIB-2 in the presence of an excess of ethylene (2 equivalents) at 30 bar and 370 °C (eqn (2), Scheme 1) and MgO is used to catalyse the isomerisation of DIB-1 to DIB-2 as it gets consumed during the metathesis leading to a constant thermodynamic ratio.4 Furthermore, branched hexenes are valuable additives for gasoline. Especially 2,3-dimethylbutenes (2,3-DMBs) after a simple hydrogenation to 2,3-dimethylbutane give an impressive research octane number (RON) of 103.5 and low RVP (Reid Vapour Pressure).5 2,3-DMBs are generally prepared by dimerisation of propylene on nickel catalysts such as in the IFP DifasolTM process, which consist of a nickel salt in the presence of bulky basic phosphine and an Al-alkylating agent dissolved in ionic liquid.6 However, these processes are dependent on ethylene or propylene, which have in recent years become desired starting materials, resulting in a notable increase in the price.7 Consequently, the development of a new approach to obtain 2,3-DMBs and neohexene (isomers easy to separate by reactive distillation as in the Sumitomo process)8 directly from alternative feeds, for example the less expensive and readily available isobutene, is highly desirable. Up to now, no direct process for the production of 2,3-DMBs and neohexene starting from isobutene has been developed. The major challenge for this approach is to identify a series of catalysts that are compatible and suitable for dimerisation and metathesis reactions under the same reaction conditions. For instance, dimerisation catalysts are known to work at low temperature to avoid cracking whereas the olefin metathesis catalyst WO3/SiO2 requires high temperature (>350 °C) to be efficient.6 However, it was demonstrated that homogenous catalysts can reach similar or better catalytic performances than the classic Phillips triolefin catalyst (WO3/SiO2) at temperature below 100 °C.9,10
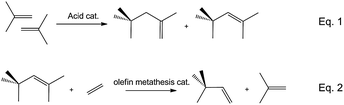 | ||
| Scheme 1 Neohexene production. | ||
When considering the design of single-site heterogeneous catalysts with a controlled coordination sphere (as homogeneous catalyst), surface organometallic chemistry (SOMC) has emerged as a powerful approach.11 In particular, we reported a tungsten based catalyst, W–H/Al2O3-(500), which gave higher turnover numbers (T.O.N.) in alkenes metathesis than the Phillips triolefin catalyst for propene production from butenes.12,13 Moreover, tungsten hydride supported on alumina has been demonstrated to be capable of catalysing refractory reactions such as non-oxidative coupling of methane,14 low temperature wax hydrogenolysis15 and direct conversion of ethylene to propylene.16
In addition, we recently reported the unique reactivity of W–H/Al2O3-(500) with 2-methylpropane to yield 2,3-dimethylbutane and ethane (eqn (3), Scheme 2).17 This reaction occurs on a single site metal–carbene-hydride, M((![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHR)(H)), and undergoes: (i) dehydrogenation of isobutane to isobutene via C–H activation on metal hydride (M–H), (ii) isobutene metathesis to 2,3-dimethylbutene and ethylene on the carbene moiety (MCHR), and (iii) hydrogenation on metal hydride (M–H) of the formed olefins.17 This result shows that supported tungsten hydride is active in the conversion of isobutene to 2,3-dimethylbutenes and ethylene.
CHR)(H)), and undergoes: (i) dehydrogenation of isobutane to isobutene via C–H activation on metal hydride (M–H), (ii) isobutene metathesis to 2,3-dimethylbutene and ethylene on the carbene moiety (MCHR), and (iii) hydrogenation on metal hydride (M–H) of the formed olefins.17 This result shows that supported tungsten hydride is active in the conversion of isobutene to 2,3-dimethylbutenes and ethylene.
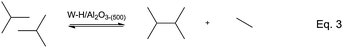 | ||
| Scheme 2 2-Methylpropane self-metathesis. | ||
In fact, when isobutene self-metathesis is performed in a dynamic flow reactor (W = 3.86 wt%, T = 150 °C, P = 1 bar and flow rate = 4 ml min−1), the reaction gives an initial maximal conversion of 35% before reaching a pseudo-plateau of 13% (Fig. S1, ESI†). The product selectivities at the steady-state are 54% branched hexenes, 30% ethylene, 12% diisobutenes (DIB-1/DIB-2 = 3), 3% isopentene and 1% propylene. The main hexene components are DMBs with high relative selectivity (92%, DMB-2/DMB-1 = 4) obtained by self-metathesis of isobutene accompanied with unexpected neohexene 8% (Fig. S2, ESI†). Formation of DMB-1 results from DMB-2 isomerisation reaction (eqn (S1), ESI†) whereas DIB-1 and DIB-2 arise from methylpropene dimerisation (eqn (1), Scheme 1) on the acidic sites of alumina. These results are confirmed by a blank reaction with alumina alone under the same reaction conditions (Fig. S3, ESI†). We observe that the conversion of isobutene is high at the beginning (14%) and decreases with time on stream to reach 2% giving selectively DIBs (DIB-1/DIB-2 = 4, see Fig. S3, ESI†). The observed deactivation is probably due to the formation of coke.18
Simultaneously, the formation of neohexene is explained by the cross metathesis reaction between DIB-2 and the in situ formed ethylene catalysed by the tungsten carbene moiety (eqn (2), Scheme 1). To confirm the latter hypothesis, a (5/95) mixture of DIB-2 and ethylene is passed over a catalytic bed of supported tungsten hydride (W = 3.86 wt%, T = 150 °C, P = 1 bar and flow rate = 4 ml min−1) at 150 °C; a high conversion (82%) of DIB-2 is obtained before reaching a pseudo-plateau of 42% (Fig. S5, ESI†). The product selectivities due to cross-metathesis reaction between DIB and ethylene at the pseudo steady-state were 44% neohexene, 53% isobutene and 3% DMBs (Fig. S6, ESI†). Hence, the low relative neohexene selectivity (8%) obtained from isobutene feed on supported W–H/Al2O3-(500) (Fig. 2b) most likely originates from the decrease in DIB production on acidic alumina sites. So far, the multifunctional supported W–H catalyst has shown the ability to form neohexene directly from pure isobutene without external addition of ethylene in the feed. However, due to the rather fast deactivation of the acidic dimerisation site, the overall selectivity in neohexene is inevitably limited. Following these preliminary studies we were interested in developing another active phase to increase the yield of valuable branched hexene products and in particular neohexene. An important criterion is that the dimerisation and olefin metathesis active phases have to be efficient at the same working temperature range (150 °C).19
This prompted us to investigate a nickel based system as previous studies have demonstrated that the nickel catalyst supported on alumina can selectively perform olefin dimerisation.20 The authors claim that the presence of Ni2+ is clearly responsible for the high selectivity of the dimers. The nickel cations show a great capacity to coordinate the π-allyl electrons of the olefins, avoiding in this way the successive addition of isobutene yielding selectively DIB.21 The association of this catalytic phase along with a proximate tungsten hydride centre may both allow the dimerisation reaction as well as the cross metathesis reaction. The modified support Ni1%–Al2O3-(500) was prepared by impregnation of a calcinated alumina powder (γ-alumina obtained from Degussa with a specific area of 100 m2 g−1) with an aqueous solution of nickel sulphate hexahydrate at 25 °C under air for 1 hour in order to load 1 wt% of nickel. The solution was evaporated under vacuum before extended drying overnight at 110 °C. Finally, the powder was calcined at 500 °C under a dry air flow for 12 h before reduction of nickel at 390 °C under hydrogen for 20 h and evacuation under high vacuum at 500 °C for 2 h to remove chemisorbed hydrogen.20 After loading the alumina surface with 1 wt% nickel, the tungsten hydride has been generated following the same protocol used for pure alumina. The procedure consists of the reaction of [W(![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CtBu)(CH2tBu)3] with the remaining surface Al–OH group (Fig. S7, ESI†) of the Ni1%–Al2O3-(500) support to give a monopodal surface species in agreement with mass balance analysis and infrared spectroscopy (ESI†). The resulting perhydrocarbyl surface species is then treated at 150 °C in the presence of anhydrous H2 (550 Torr) for 15 h to give the corresponding hydride along with 14.5 methane per tungsten atom (determined by GC).22 Infrared spectroscopy reveals the disappearance of the alkyl groups and appearance of the same hydride stretching bands at 1910 and 1800 cm−1 as observed with the W–H/Al2O3-(500) system (Fig. S7, ESI†).21 A resonance at 1370 cm−1 corresponding to the SO bands is indicative of the remaining sulphate group. The elemental analysis results show a lower tungsten loading (2.2 wt%) on the nickel modified support in comparison to the native Al2O3-(500). This can be explained by a partial hindering of the aluminoxy group by the nickel sulphate.
CtBu)(CH2tBu)3] with the remaining surface Al–OH group (Fig. S7, ESI†) of the Ni1%–Al2O3-(500) support to give a monopodal surface species in agreement with mass balance analysis and infrared spectroscopy (ESI†). The resulting perhydrocarbyl surface species is then treated at 150 °C in the presence of anhydrous H2 (550 Torr) for 15 h to give the corresponding hydride along with 14.5 methane per tungsten atom (determined by GC).22 Infrared spectroscopy reveals the disappearance of the alkyl groups and appearance of the same hydride stretching bands at 1910 and 1800 cm−1 as observed with the W–H/Al2O3-(500) system (Fig. S7, ESI†).21 A resonance at 1370 cm−1 corresponding to the SO bands is indicative of the remaining sulphate group. The elemental analysis results show a lower tungsten loading (2.2 wt%) on the nickel modified support in comparison to the native Al2O3-(500). This can be explained by a partial hindering of the aluminoxy group by the nickel sulphate.
When isobutene is flowed through the nickel-promoted alumina-supported tungsten hydride, W–H/Ni1%–Al2O3-(500) (W = 2.2 wt%, T = 150 °C, P = 1 bar and flow rate 4 ml min−1), the conversion reaches a maximum of 27% and stabilises at 14% to give a T.O.N of 320 after 16 hours (Fig. 1a). The observed activity is 1.37 fold higher than that obtained with W–H/Al2O3-(500) (T.O.N of 230). At the pseudo-steady state, the observed selectivities in isobutene conversion are: 68.1% branched hexenes, (compared to 54% with W–H/Al2O3-(500)), 12.5% ethylene, 11.4% (DIB), 5.5% 3-methylbut-1-ene and finally 2.2% propylene (Fig. 1b).
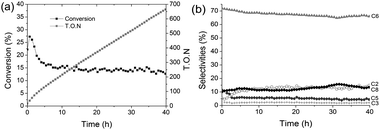 | ||
Fig. 1 Conversion of isobutene catalysed by W–H/Ni1%–Al2O3-(500) (2.2 wt% W): (a) conversion of isobutene (■) and T.O.N ( ); (b) selectivities: ( ); (b) selectivities: ( ) branched hexenes; (◆) DIB; (○) ethylene; ( ) branched hexenes; (◆) DIB; (○) ethylene; ( ) 3-methylbut-1-ene; (+) propylene. ) 3-methylbut-1-ene; (+) propylene. | ||
Noteworthy, the relative selectivity between the branched hexene isomers shows a 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :40 distribution between 2,3-DMBs (DMB-2/DMB-1 = 3) and neohexene (Fig. 2a). As in the case of the catalysis performed with W–H/Al2O3-(500), 2,3-DMBs are produced by the productive self-metathesis of isobutene involving an unfavoured 1,2-disubstituted metallacycle intermediate 5a (Scheme 3). It is important to notice that two other intermediates could be formed from the reverse favoured approach of isobutene on 4a and 4b, but they lead to degenerate metathesis. Productive metathesis of isobutene involving 5a is thus disfavoured for steric reasons which accounts for the relatively moderate activity observed in the conversion of isobutene in comparison to linear alkenes.11,12 The comparison of the hexenes relative selectivity between the native catalyst (Fig. 2b) and the nickel modified catalyst, W–H/Ni1%–Al2O3-(500), shows the beneficial effect of the promoter on the neohexene selectivity (40% in comparison to W–H/Al2O3-(500) 8%), which is indeed higher and, more importantly, stays stable with time on stream, in contrast to the pristine alumina supported W–H (Fig. 2a). These results along with the low selectivity in ethylene demonstrate the ability of the W–H/Ni1%–Al2O3-(500) catalyst to perform both the self-metathesis and the selective dimerisation of isobutene to DIB followed by cross metathesis between DIB and ethylene produced in situ. These results highlight the importance of the dual functionality of the new supported W–H/Ni1%–Al2O3-(500) catalyst in the direct conversion of isobutene into DMBs and neohexene with improved selectivity.23 In addition to isobutene dimerisation reaction giving DIB (eqn (1)), a catalytic cycle, that involves isobutene self-metathesis to DMB, and DIB/ethylene cross-metathesis, can be proposed (Scheme 3).
:40 distribution between 2,3-DMBs (DMB-2/DMB-1 = 3) and neohexene (Fig. 2a). As in the case of the catalysis performed with W–H/Al2O3-(500), 2,3-DMBs are produced by the productive self-metathesis of isobutene involving an unfavoured 1,2-disubstituted metallacycle intermediate 5a (Scheme 3). It is important to notice that two other intermediates could be formed from the reverse favoured approach of isobutene on 4a and 4b, but they lead to degenerate metathesis. Productive metathesis of isobutene involving 5a is thus disfavoured for steric reasons which accounts for the relatively moderate activity observed in the conversion of isobutene in comparison to linear alkenes.11,12 The comparison of the hexenes relative selectivity between the native catalyst (Fig. 2b) and the nickel modified catalyst, W–H/Ni1%–Al2O3-(500), shows the beneficial effect of the promoter on the neohexene selectivity (40% in comparison to W–H/Al2O3-(500) 8%), which is indeed higher and, more importantly, stays stable with time on stream, in contrast to the pristine alumina supported W–H (Fig. 2a). These results along with the low selectivity in ethylene demonstrate the ability of the W–H/Ni1%–Al2O3-(500) catalyst to perform both the self-metathesis and the selective dimerisation of isobutene to DIB followed by cross metathesis between DIB and ethylene produced in situ. These results highlight the importance of the dual functionality of the new supported W–H/Ni1%–Al2O3-(500) catalyst in the direct conversion of isobutene into DMBs and neohexene with improved selectivity.23 In addition to isobutene dimerisation reaction giving DIB (eqn (1)), a catalytic cycle, that involves isobutene self-metathesis to DMB, and DIB/ethylene cross-metathesis, can be proposed (Scheme 3).
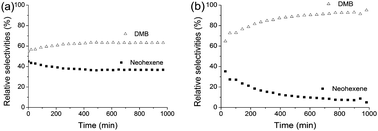 | ||
| Fig. 2 DMB (Δ) and neohexene (■) relative selectivities; (a) during the conversion of isobutene catalysed by W–H/Ni1%–Al2O3-(500); (b) during the conversion of isobutene catalysed by W–H/Al2O3-(500). | ||
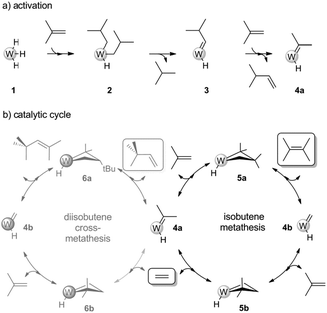 | ||
| Scheme 3 Mechanism of isobutene conversion. | ||
In conclusion, the modification of the alumina support of W–H/Al2O3-(500) with 1 wt% of nickel enhances the productivity and the selectivity in valuable branched hexenes. Current results show that branched hexenes can be readily produced in notable yield through a simple fixed bed reactor starting from pure isobutene. This methodology facilitates the existing technology that involves a multiple reactor system and applies a cheaper gas feed (isobutene alone versus ethylene–isobutene (Phillips process) and propylene (Difasol process)). Finally, the observed branched hexenes (DMB-1, DMB-2 and neohexene) can be easily separated by modern distillation technology and can thereby provide important compounds for fine and petrochemical industries.
Notes and references
- H. Sato, H. Tojima and K. Ikimi, J. Mol. Catal. A: Chem., 1999, 144, 285–293 CrossRef CAS.
- H. Mimoun, Chimia, 1996, 50, 620–625 CAS.
- R. L. Banks, D. S. Banaslak, P. S. Hudson and J. R. Norell, J. Mol. Catal., 1982, 15, 21–33 CrossRef CAS.
- J. C. Mol, J. Mol. Catal., 2004, 213, 39–45 CrossRef CAS.
- J. Hancsók, S. Magyar, K. V. S. Nguyen, L. Keresztúry and L. Valkai, Pet. Coal, 2005, 45, 99 Search PubMed.
- H. Olivier-Bourbigou, L. Magna and D. Morvan, Appl. Catal., A, 2010, 373, 1–56 CrossRef CAS.
- J. S. Plotkin, Catal. Today, 2005, 106, 10–14 CrossRef CAS.
- A. Forestière, H. Olivier-Bourbigou and L. Saussine, Oil Gas Sci. Technol., 2009, 64, 649–667 CrossRef.
- D. Astruc, New J. Chem., 2005, 29, 42–56 RSC.
- C. van Schalkwyk, A. Spamer, D. J. Moodley, T. Dube, J. Reynhardt, J. M. Botha and H. C. M. Vosloo, Appl. Catal., A, 2003, 255, 143–152 CrossRef CAS.
- E. Mazoyer, N. Merle, A. de Mallmann, J. M. Basset, E. Berrier, L. Delevoye, J. F. Paul, C. P. Nicholas, R. M. Gauvin and M. Taoufik, Chem. Commun., 2010, 46, 8944–8946 RSC.
- E. Mazoyer, K. C. Szeto, S. Norsic, A. Garron, J.-M. Basset, C. P. Nicholas and M. Taoufik, ACS Catal., 2011, 1, 1643–1646 CrossRef CAS.
- E. Mazoyer, K. C. Szeto, J.-M. Basset, C. P. Nicholas and M. Taoufik, Chem. Commun., 2012, 48, 3611–3613 RSC.
- K. C. Szeto, S. Norsic, L. Hardou, E. Le Roux, S. Chakka, J. Thivolle-Cazat, A. Baudouin, C. Papaioannou, J. M. Basset and M. Taoufik, Chem. Commun., 2010, 46, 3985–3987 RSC.
- S. Norsic, C. Larabi, M. Delgado, A. Garron, A. de Mallmann, C. Santini, K. C. Szeto, J.-M. Basset and M. Taoufik, Catal. Sci. Technol., 2012, 2, 215–219 CAS.
- M. Taoufik, E. Le Roux, J. Thivolle-Cazat and J. M. Basset, Angew. Chem., Int. Ed., 2007, 46, 7202–7205 CrossRef CAS.
- N. Merle, F. Stoffelbach, M. Taoufik, E. Le Roux, J. Thivolle-Cazat and J. M. Basset, Chem. Commun., 2009, 2523–2525 RSC.
- M. Trombetta, G. Busca, S. A. Rossini, V. Piccoli and U. Cornaro, J. Catal., 1997, 168, 334–348 CrossRef CAS.
- I. Bucsi and G. A. Olah, J. Catal., 1992, 137, 12–21 CrossRef CAS.
- Y. Chauvin, D. Commereuc, F. Hugues and J. Thivolle-cazat, Appl. Catal., A, 1988, 42, 205–216 CrossRef CAS.
- F. J. Tzompantzi, M. E. Manríquez, J. M. Padilla, G. Del Angel, R. Gómez and A. Mantilla, Catal. Today, 2008, 133–135, 154–159 CrossRef CAS.
- M. Taoufik, E. Roux, J. Thivolle-Cazat, C. Copéret, J.-M. Basset, B. Maunders and G. Sunley, Top. Catal., 2006, 40, 65–70 CrossRef CAS.
- J.-M. Basset, N. Merle, F. Stoffelbach, M. Taoufik and J. Thivolle-Cazat, Patent application, WO2009044107, 2009 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Preparation, characterisation of catalysts and catalytic test procedure. See DOI: 10.1039/c2cy20539d |
| ‡ Present Address: UPMC, Université Paris 6, CNRS, Laboratoire de Chimie des Polymères, UMR 7610, 3, rue Galilée 94200 Ivry sur Seine, France. |
| § Present Address: KAUST Catalyst Research Center, 4700 King Abdullah University of Science and Technology, Thuwal 23955-6900, Kingdom of Saudi Arabia. |
| This journal is © The Royal Society of Chemistry 2012 |