The oxidation of alcohols with trichloroisocyanuric acid: pyridine from base to organocatalyst
Ruben P.
van Summeren
a,
Amy
Romaniuk
b,
Edwin G.
IJpeij
a and
Paul L.
Alsters
*a
aDSM Innovative Synthesis B.V., PO Box 18, Geleen, 6160 MD, The Netherlands. E-mail: paul.alsters@dsm.com; Tel: +31 46 4761348
bDepartment of Pure and Applied Chemistry, University of Strathclyde, Thomas Graham Building, 295 Cathedral Street, Glasgow, Gl 1XL, UK
First published on 2nd August 2012
Abstract
An alcohol oxidation method based on the use of pyridine or 3-cyanopyridine as an organocatalyst in combination with trichloroisocyanuric acid as the terminal oxidant is reported. It is demonstrated that secondary aliphatic alcohols can be selectively oxidized to the corresponding ketones in the presence of primary aliphatic alcohols. The method is also suitable for the oxidation of primary and secondary benzylic alcohols. N-Chloropyridinium cyanurates are proposed as the active alcohol-oxidizing species.
Introduction
The oxidation of alcohols to the corresponding carbonyl compounds is one of the key reactions in synthetic organic chemistry. Consequently, a wide range of methods has been developed over the years to address this transformation.1 Traditionally, stoichiometric approaches are used because of their high selectivities and yields. These include such well-known methods like Swern or Oppenauer oxidations and oxidations with chromium(VI) or hypervalent iodine reagents.2 However, environmental- and health considerations inspired the discovery of alternative processes. More recently, catalytic methods based on transition metals or organocatalysts3 and also biocatalytic methods4 have been reported in combination with various terminal oxidants. Nevertheless, despite this plethora of methods to choose from, alcohol oxidation is still considered to be a difficult step on an industrial scale. This is mainly due to the large number of issues that have to be simultaneously solved to make a viable process. To name but a few: safety, cost, toxicity, environmental impact, (chemo)-selectivity and sustainability all need to be adequately tackled. Hence, depending on the exact conversion and product application at hand, a suitable method may or may not be available off the shelf. Accordingly, alternative scalable alcohol oxidation methods remain highly sought after.In 1992, Hiegel and Nalbandy disclosed a particularly practical method for the conversion of secondary alcohols into ketones.5 Specifically, they demonstrated that a range of secondary alcohols can be oxidized at ambient temperature by treatment with trichloroisocyanuric acid (TCCA) in acetone in the presence of a slight excess of pyridine as a base (Scheme 1). Furthermore it was shown that primary alcohols are oxidized at a significantly lower rate, allowing for chemoselective oxidations without the need for protecting groups.
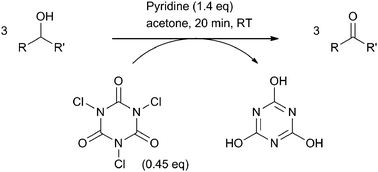 | ||
| Scheme 1 Secondary alcohol oxidation with trichloroisocyanuric acid in the presence of pyridine.5 | ||
In principle, the method as described by Hiegel and Nalbandy is an attractive candidate for scale-up. The procedure is operationally simple and the reaction conditions are relatively mild. Moreover, TCCA is readily available, easy to handle, inexpensive and has a high active chlorine content. The only drawback is the use of stoichiometric amounts of pyridine which is a CMR (Carcinogenic/Mutagenic/Reprotoxic) category 3 compound. Consequently, we were interested in further optimization of the reaction conditions including a screening for alternative solvent-base combinations, aiming at reducing the required amount of pyridine. However, it soon became apparent (vide infra) that the role of pyridine in the reaction is much larger than just a scavenger for the hydrochloric acid that is generated during alcohol oxidation and that, in fact, under proper conditions pyridine acts as an organocatalyst for the alcohol oxidation.
Experimental
General: chemicals were obtained from commercial sources and used without further purification. 1H-NMR spectra were recorded on a Bruker Avance 300 MHz spectrometer. GC analysis was performed on a 5890 Series II Gas Chromatographer with a CP-Sil 5 CB 25 × 0.32 (1.2) column.General method using stoichiometric pyridine: Method A – the alcohol (2.5 mmol, 1.0 eq.) was dissolved in deuterated acetonitrile (2.5 mL) and stirred under a nitrogen atmosphere. An ambient temperature was maintained by external cooling using a water bath. Subsequently, pyridine (0.25 mL, 3.1 mmol, 1.2 eq.) was added after which a solution of TCCA (0.25 g, 1.1 mmol, 0.43 eq.) in deuterated acetonitrile (2.5 mL) was gradually added over 30 minutes using a syringe pump. The resulting mixture was stirred for the appropriate reaction time at which point neopentyl bromide (0.196 g, 1.30 mmol) was added as an internal standard. A sample was then filtered and analyzed by 1H-NMR.
General method using catalytic pyridine: Method B – sodium bicarbonate (0.25 g, 3.0 mmol, 1.2 eq.) was suspended in DMF and stirred for 30 minutes under a nitrogen atmosphere. An ambient temperature was maintained by external cooling using a water bath. Subsequently, the alcohol (2.5 mmol, 1.0 eq.) and pyridine (30 μL, 0.37 mmol, 15 mol%) were added. Next a solution of TCCA (0.25 g, 1.1 mmol, 0.43 eq.) in DMF (2.5 mL) was gradually added over 30 minutes using a syringe pump. The resulting mixture was stirred for the appropriate reaction time at which point 1,2-dichlorobenzene (0.156 g, 1.06 mmol) was added as an internal standard. A sample was then filtered, diluted with isopropanol and analyzed by GC.
Results and discussion
The investigation started with solvent screening using menthol as a model substrate (Table 1). Standard reaction conditions entailed the use of a small excess of TCCA (1.05 eq. positive chlorine) in the presence of stoichiometric pyridine. Under these circumstances, the selectivity towards menthone was high for all solvents tested with slightly diminished values for solvents containing an ester moiety (entries 3 and 4). Most importantly, no chlorinated side products were detected (GC). In contrast, the activity of the system proved to be strongly solvent dependent. The best results were obtained in acetonitrile and acetone (entries 1 and 9) with virtually complete conversion in 20 minutes. In MTBE on the other hand, only 14% conversion was achieved after the same time span (entry 2). This difference is most likely due to the poor solubility of TCCA and related partly chlorinated isocyanuric acid intermediates in apolar solvents. Based on these results, acetonitrile was selected as the solvent of choice for the subsequent base screening.| Entry | Solvent | Conv.b | Conv.b | Sel.b |
|---|---|---|---|---|
| 20 min (%) | 120 min (%) | 120 min (%) | ||
| a Conditions: menthol (5 mmol), solvent (10 mL), pyridine (5 mmol), TCCA (1.75 mmol), RT. b Conversion and selectivity towards menthone determined by quantitative GC with 1,2-dichlorobenzene as internal standard. c Accurate determination hampered by the presence of undissolved TCCA. | ||||
| 1 | Acetone | 97 | 97 | >99 |
| 2 | MTBE | n.d.c | 74 | >99 |
| 3 | Ethylacetate | 59 | 95 | 94 |
| 4 | n-Butylacetate | 42 | 78 | 84 |
| 5 | Dimethylcarbonate | 58 | 87 | >99 |
| 6 | Dichloromethane | 67 | 79 | >99 |
| 7 | 1,2-Dichloroethane | n.d.c | 65 | >99 |
| 8 | Chloroform | n.d.c | 68 | >99 |
| 9 | Acetonitrile | 98 | 100 | >99 |
Hiegel and Nalbandy reported that the desired alcohol oxidation reaction also occurs under acidic conditions albeit with lower chemoselectivity.6 The primary role of pyridine is therefore to increase selectivity by trapping hydrogen chloride. If this line of reasoning is correct, it should be straightforward to replace pyridine by an alternative base. Hence, a number of organic and inorganic bases were evaluated for this purpose, using the oxidation of menthol as a test reaction (Table 2). Surprisingly, most bases had a negative effect on both the activity and especially the selectivity of the oxidation system as compared to the blanc reaction (entry 1 vs. entries 3–11). It appeared that a.o. triethylamine, imidazole, aniline, DABCO, sodium bicarbonate and N,N-dimethylaniline block (to some extend) successfully the unselective alcohol oxidation reaction that occurs under acidic conditions. However, unlike pyridine, these bases unfortunately do not facilitate an alternative, more selective pathway towards the desired carbonyl compound. Instead, GC-MS analysis indicated that unselective chlorination processes prevented a clean alcohol oxidation. Only cesium carbonate (entry 13) and the pyridine derivative 2,6-lutidine (entry 14) exhibited similar selectivity as pyridine (entry 2), although with significantly lower conversions. Consequently, it should be concluded that in the present context pyridine is a privileged compound whose modus operandi surpasses the simple action of a base.
| Entry | Base | Conv.b (%) | Sel.b (%) |
|---|---|---|---|
| a Conditions: menthol (5 mmol), CH3CN (10 mL), base (5 mmol), TCCA (1.75 mmol), RT, 20 minutes. b Conversion and selectivity towards menthone determined by quantitative GC with 1,2-dichlorobenzene as internal standard. | |||
| 1 | None | 50 | 81 |
| 2 | Pyridine | 100 | >99 |
| 3 | Pyrazine | 70 | 65 |
| 4 | N,N-Dimethylaminopyridine | 24 | <1 |
| 5 | Triethylamine | 47 | 2 |
| 6 | Imidazole | 26 | 1 |
| 7 | N,N-Dimethylaniline | 17 | 5 |
| 8 | Aniline | 17 | 0 |
| 9 | Sodium bicarbonate | 62 | 27 |
| 10 | 1,4-Diazabicyclo[2.2.2]octane (DABCO) | 26 | 9 |
| 11 | 2,6-Di-tert-butylpyridine | 95 | 13 |
| 12 | Potassium acetate | 27 | 86 |
| 13 | Cesium carbonate | 52 | >99 |
| 14 | 2,6-Lutidine | 37 | 95 |
When the reaction was performed in the presence of pyridine, it was observed that the initially homogeneous reaction mixture temporarily turned into a white suspension. This suspension subsequently became a clear solution again before cyanuric acid finally precipitated at the end of the reaction. We were intrigued by the nature of the initially formed precipitate and suspected that this was a reactive, poorly soluble intermediate generated from pyridine and TCCA. Upon closer inspection in the absence of substrate, it was indeed found that addition of 3 mol equivalents of pyridine to a CD3CN solution of TCCA (i.e. 1/1 py/Cl molar ratio) at room temperature results in the gradual precipitation of a white, slightly yellowish solid. 1H-NMR analysis in D2O of three batches of this solid, isolated by decantation of the CD3CN supernatant7 and drying of the solid in a stream of N2, showed the characteristic doublet/triplet/triplet pattern with a 2/1/2 integral ratio of a pyridine-type structure, but significantly shifted down-field relative to pyridine itself.8 Although the chemical shifts of these three batches relative to the HDO peak set at 4.70 ppm varied slightly, they were in all cases clearly distinct from those measured in D2O for pyridinium chloride and pyridine N-oxide, these compounds being two other potential solid products generated from pyridine + TCCA.9 The presence of a pyridine-type structure in the precipitate that is distinct from that in pyridinium chloride and pyridine N-oxide was further confirmed by 13C-NMR in D2O.10 We feel the foregoing results are in line with chlorine transfer from TCCA to pyridine, generating poorly soluble tris[N-chloropyridinium] cyanurate (Scheme 2).11,12
![Formation of tris[N-chloropyridinium] cyanurate by positive chlorine transfer from TCCA to pyridine.](/image/article/2012/CY/c2cy20403g/c2cy20403g-s2.gif) | ||
| Scheme 2 Formation of tris[N-chloropyridinium] cyanurate by positive chlorine transfer from TCCA to pyridine. | ||
The results above illustrate that pyridine + TCCA, through the formation of N-chloropyridinium cyanurate species, acts as a unique alcohol oxidation system, with the cyanurate anion acting as a hydroxyl proton acceptor and the N-chloropyridinium cation acting as an α-CH bond hydride abstractor, as depicted in Scheme 3. Such a hydride abstraction mechanism is in line with the fact that secondary alcohols are oxidized faster than primary alcohols (see also entry 2 of Table 4) as it involves a transient positive charge on the carbon atom that is being oxidized. Further (circumstantial) evidence is provided by a direct competition experiment between electron rich ortho-methylbenzylalcohol and electron poor ortho-chlorobenzylalcohol using limiting oxidants. Specifically, it was found that the former substrate is oxidized substantially faster resulting in a mixture of the corresponding aldehydes with a ratio of 88![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :12 (data not shown). Mechanistically, the pyridine + TCCA system most likely resembles other reported alcohol oxidation systems based on cationic reagents comprising halogen cations stabilized by neutral organic nitrogen bases.13
:12 (data not shown). Mechanistically, the pyridine + TCCA system most likely resembles other reported alcohol oxidation systems based on cationic reagents comprising halogen cations stabilized by neutral organic nitrogen bases.13
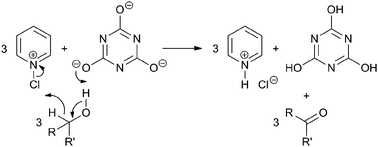 | ||
| Scheme 3 Proposed alcohol oxidation mechanism based on hydride abstraction by N-chloropyridinium cations. | ||
In view of the distinctive role of pyridine in the reaction, efforts were shifted from replacing pyridine altogether to reducing the amount of pyridine to catalytic proportions. In particular, it was envisioned that pyridine could operate as an organocatalyst in conjunction with a cheap and non-toxic base that merely acts as a hydrochloric acid scavenger. To this end, a variety of bases were examined in different solvents (Table 3). It was soon established that catalytic pyridine did not have any beneficial effect when combined with potassium acetate, DABCO, triethylamine or cesium carbonate using acetonitrile as the solvent (entries 1–4 in Table 3vs. entries 5, 10, 12 and 13 in Table 2). However, the reaction outcome did substantially improve in the case of sodium bicarbonate. Specifically, the GC-yield increased from 17% to 39% due to both higher conversion as well as enhanced selectivity (entry 6 in Table 3vs. entry 9 in Table 2). At this point, it was reasoned that the availability of dissolved sodium bicarbonate most likely constituted a limiting factor in the reaction. To remove this bottleneck, co-solvents such as methanol, 1,2-dichloroethane and methyl–isobutyl ketone were tested but unfortunately to no avail (data not shown). On the other hand, when the reaction was performed in DMF with slow addition of the oxidant and only 1 eq. NaHCO3, the conversion and selectivity indeed improved to a gratifying 96% and 85% respectively (entry 8). The favorable influence of pyridine under these conditions was clearly established in the control experiment without organocatalyst (entry 9).
| Entry | Base | Solvent | Conv.b (%) | Sel.b (%) |
|---|---|---|---|---|
| a Conditions: menthol (5 mmol), solvent (10 mL), pyridine (0.5 mmol, 10 mol%), base (1 or 2 eq.), TCCA (1.75 mmol), RT, 2 hours. b Conversion and selectivity towards menthone determined by quantitative GC with 1,2-dichlorobenzene as internal standard. c TCCA in 5 mL DMF dosed over 1 h to 5 mL reaction mixture. d Without pyridine. | ||||
| 1 | Potassium acetate (2 eq.) | CH3CN | 67 | 9 |
| 2 | DABCO (1 eq.) | CH3CN | <5 | — |
| 3 | Triethylamine (1 eq.) | CH3CN | 22 | <5 |
| 4 | Cesium carbonate (1 eq.) | CH3CN | 60 | 95 |
| 5 | Sodium carbonate (2 eq.) | CH3CN | <5 | — |
| 6 | Sodium bicarbonate (2 eq.) | CH3CN | 80 | 49 |
| 7 | Sodium bicarbonate (1 eq.) | DMF | 86 | 25 |
| 8c | Sodium bicarbonate (1 eq.) | DMF | 96 | 85 |
| 9c,d | Sodium bicarbonate (1 eq.) | DMF | 76 | 70 |
With a proof-of-principle for the organocatalytic oxidation of cyclic alcohols in hand, the next step was to determine the substrate scope in comparison to the original procedure using stoichiometric pyridine. As can be seen in Table 4, also linear aliphatic secondary alcohols are cleanly oxidized to the corresponding ketones by both methods (entry 1). It should be noted though that the catalytic protocol is considerably slower resulting in a modest 50% conversion after 24 hours. Furthermore, oxidation of 1,3-butanediol clearly demonstrated that secondary aliphatic alcohol groups are generally favored over primary ones (entry 2).14 In this case, both the activity as well as the selectivity of the procedure with stoichiometric pyridine are superior. In contrast, primary and secondary benzylic alcohols are effectively converted by either protocol to the corresponding aldehyde or ketone, respectively (entries 3 and 4). A competition experiment between benzylalcohol and 1-phenyl ethanol revealed that also for benzylic substrates the secondary alcohol is slightly preferred although the distinction is much less pronounced in this case (factor of only 2). Interestingly, neither method proved useful for the preparation of anisaldehyde (entry 5). Apparently, electron rich benzylic alcohols are unsuitable substrates due to the susceptibility of the aromatic ring towards degradative ring chlorination (GC-MS evidence).15 Electron poor benzylic substrates on the other hand are cleanly oxidized, albeit at a reduced rate leading to several days of reaction time (entries 6 and 7). Overall, it can be concluded that the oxidation protocol with catalytic pyridine follows the same trends as its stoichiometric counterpart but with a somewhat inferior activity/selectivity profile.
| Entry | Substrate | Method A (stoichiometric) | Method B (catalytic) |
|---|---|---|---|
| Time, conv. (%), sel.b (%) | Time, conv. (%), sel.b (%) | ||
| a Conditions: Method A: substrate (2.5 mmol), CD3CN (2.5 mL), pyridine (3.1 mmol), TCCA (1.1 mmol) in 2.5 mL CD3CN dosed over 30 min, RT; Method B: substrate (2.5 mmol), DMF (2.5 mL), pyridine (0.38 mmol, 15 mol%), NaHCO3 (3.13 mmol), TCCA (1.05 mmol) in 2.5 mL DMF dosed over 30 min, RT. b Selectivity towards the corresponding ketone or aldehyde. c Determined by quantitative 1H-NMR with neopentyl bromide as internal standard. d Determined by quantitative GC with 1,2-dichlorobenzene as internal standard. e Selectivity towards 4-hydroxy-2-butanone. f DMF-d7 used. g See the text: degradative ring chlorination. | |||
| 1 |

|
1 hour, 100c, 97c | 24 hours, 50d, >99d |
| 2 |

|
1 hour, 90c, >95c,e | 6 hours, 54c,f, >95c,e,f |
| 3 |

|
4 hours, 100d, 96d | 6 hours, 98d, 91d |
| 4 |

|
4 hours, 97c, 93c | 5.5 hours, 91d, 87d |
| 5 |
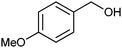
|
4 hours, 55c, 0c,g | 6 hours, 73d, 0d,g |
| 6 |

|
16 hours, 70c, >99c | 3 days, 70d, >99d |
| 7 |

|
2 days, 18c, 98c | 2 days, 31d, 92d |
Finally, in an attempt to find an organocatalyst with a better activity–selectivity profile, three alternative pyridine derivatives were briefly assessed in the oxidation of 2-octanol, using conditions as described under Method B in Table 4. It was found that para-methoxy pyridine performed somewhat less than pyridine itself giving the desired ketone with 44% conversion and 90% selectivity after 24 hours. Surprisingly, full conversion was achieved with 4-cyano-pyridine but unfortunately at the cost of a deteriorated selectivity (69%). However, 3-cyano-pyridine displayed a twice as high activity as pyridine while being equally selective, leading to an overall yield of 94% of 2-octanone.
Conclusions
The pyridine + TCCA mediated oxidation of alcohols to the corresponding carbonyl compounds is an interesting candidate for application on an industrial scale. The present work focused on substituting pyridine with a cheap and benign alternative. Unexpectedly, it was found that the role of pyridine in fact goes far beyond the simple task of a base. In particular, evidence has been provided that the unique combination of pyridine and trichloroisocyanuric acid, through in situ formation of N-chloropyridinium cyanurate species, facilitates alcohol oxidation via α-CH bond hydride abstraction by the chloropyridinium cation and hydroxyl proton transfer to the cyanurate anion. Consequently, the aim of the investigation was adjusted from replacing pyridine altogether towards the use of pyridine as an organocatalyst in the presence of a stoichiometric inorganic base. In this respect it turned out that sodium bicarbonate is in principle suitable for this purpose under carefully optimized reaction conditions comprising slow addition of the oxidant and the use of DMF as solvent. Although the use of DMF is disadvantageous in the organocatalytic protocol, development of the latter did allow meeting the target of strongly reducing the required amount of pyridine. We demonstrated that the activity of the organocatalytic system can be enhanced by the use of pyridine derivatives bearing an electron withdrawing substituent on the aromatic ring, with 3-cyanopyridine allowing rate enhancement while maintaining a high selectivity. This could serve as a basis for the identification of an advanced organocatalytic method.Acknowledgements
The authors thank Gerard Verzijl and Ben de Lange (DSM Innovative Synthesis B.V.) for the very helpful suggestions.Notes and references
- G. Tojo and M. Fernández, Oxidation of Alcohols to Aldehydes and Ketones. Basic Reactions in Organic Synthesis, Springer, New York, 2010 Search PubMed.
- M. Hudlický, Oxidations in Organic Chemistry, American Chemical Society, Washington, 1990 Search PubMed.
- I. W. C. E. Arends and R. A. Sheldon, in Modern Oxidation Methods, ed. J. E. Bäckvall, Wiley-VCH, Weinheim, 2010 Search PubMed.
- F. Hollmann, I. W. C. E. Arends, K. Buehler, A. Schallmey and B. Bühler, Green Chem., 2011, 13, 226–265 RSC.
- G. A. Hiegel and M. Nalbandy, Synth. Commun., 1992, 22, 1589–1595 CrossRef CAS.
- Among others, α-halogenation and degradation of TCCA into dichlorine gas occur.
- After decantation, more solid continued to precipitate gradually from the CD3CN supernatant. In this way, three batches of precipitate were isolated over time.
- 300 MHz 1H-NMR data of the pyridine + TCCA precipitate relative to the HDO peak set at 4.70 ppm: δ (ppm): 8.79–8.62 (d, 2H), 8.45–8.30 (t, 1H), 7.94–7.80 (t, 2H). The indicated chemical shift spread corresponds to the spread observed for the three batches isolated for this compound.
- Pyridinium chloride is readily soluble in both water and acetonitrile. In contrast, the pyridine + TCCA precipitate is insoluble in acetonitrile and dissolves only slowly in water. These observations further underline that the precipitate is not pyridinium chloride.
- 75 MHz 13C-NMR data (D2O solvent) of the pyridine + TCCA precipitate: δ (ppm): 146, 144, 127.
- The poor solubility of this species did not allow observation of the cyanurate carbons by 13C-NMR.
- N-Chloropyridinium salts have been prepared earlier without characterization by NMR from pyridine + Cl2 in TFA: J. R. Lindsay Smith, L. C. McKeer and J. M. Taylor, J. Chem. Soc., Perkin Trans. 2, 1987, 1533–1537 RSC.
- (a) G. Rousseau and S. Robin, Tetrahedron Lett., 2000, 41, 8881–8885 CrossRef CAS; (b) L. K. Blair, S. Hobbs, N. Bagnoli, L. Husband and N. Badika, J. Org. Chem., 1992, 57, 1600–1603 CrossRef CAS.
- Primary aliphatic alcohols are in fact oxidized to the corresponding aldehydes in the absence of a secondary alcohol. However, reaction rates were considered too low to be of practical use.
- (a) H. V. Carrillo, A. Y. Rodriguez, R. G. Landolt and W. H. Hendrickson, Synlett, 2011, 2069–2071 CAS; (b) K. V. Sarkanen and C. W. Dence, J. Org. Chem., 1960, 25, 715–720 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |