Steam reforming of ethanol at medium pressure over Ru/Al2O3: effect of temperature and catalyst deactivation
Received
27th April 2012
, Accepted 25th May 2012
First published on 15th June 2012
Abstract
In this study a low-loaded 0.2% Ru/alumina catalyst was tested over a range of temperatures (773–873 K) at 20 barg with a 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 steam to ethanol ratio. Ruthenium was found to produce significant amounts of ethene, although in the steady state hydrogen was always the main product. Significant levels of methane and carbon dioxide were formed at 873 K. Hydrogenation of ethene to ethane was inhibited and an activation energy comparable with low temperature studies was obtained. Liquid products such as acetaldehyde, diethyl ether and acetone were also produced at low levels (<12%). Temperature-programmed oxidation (TPO), Raman spectroscopy, powder XRD, BET and SEM analysis were used to characterise the nature of the coke species for reactions at 773 K, 823 K and 873 K. The TPO results indicate that different types of coke are deposited on the catalyst during ethanol steam reforming (ESR). Carbon nanotubes and filamentous coke were observed on the catalyst after ESR at 873 K. Powder XRD and Raman analysis revealed that the coke deposited on the catalysts was graphitic in nature and the disorder in the graphitic type coke generally increased with in an increase in the reaction temperature.
:1 steam to ethanol ratio. Ruthenium was found to produce significant amounts of ethene, although in the steady state hydrogen was always the main product. Significant levels of methane and carbon dioxide were formed at 873 K. Hydrogenation of ethene to ethane was inhibited and an activation energy comparable with low temperature studies was obtained. Liquid products such as acetaldehyde, diethyl ether and acetone were also produced at low levels (<12%). Temperature-programmed oxidation (TPO), Raman spectroscopy, powder XRD, BET and SEM analysis were used to characterise the nature of the coke species for reactions at 773 K, 823 K and 873 K. The TPO results indicate that different types of coke are deposited on the catalyst during ethanol steam reforming (ESR). Carbon nanotubes and filamentous coke were observed on the catalyst after ESR at 873 K. Powder XRD and Raman analysis revealed that the coke deposited on the catalysts was graphitic in nature and the disorder in the graphitic type coke generally increased with in an increase in the reaction temperature.
Introduction
Currently approximately 80% of the world energy needs are met directly or indirectly from non-renewable sources such as fossil fuels.1 The complexity associated with fossil fuel use as an energy source (pollution, global warming, finite supplies) and our subsequent dependence on them have heightened the need to find new alternative energy supplies.2 This has led a move away from fossil fuels and the development of technology using renewable feedstocks.3 Renewable energy is often considered in terms of wind, solar, and tidal but these, in general, contribute to static power generation in the form of electricity. However renewable or sustainable feedstocks relate more to the conversion of biomass to energy in the form of fuel rather than electricity (although that is perfectly feasible and may play a role in the future). There are various options for the conversion of biomass to fuels and a review of these options can be found in Zinoviev et al.3 however one route is the conversion of biomass to hydrogen.
Hydrogen is seen as a future energy vector that is carbon free and hence would not directly contribute CO2, the main cause of global warming, to the atmosphere. In addition, hydrogen is abundantly available in the universe and possesses the highest energy content per unit of weight (i.e. 120.7 kJ g−1) of any of the known fuel.4–6 Ideally if hydrogen was produced by electrolysis of water, using renewable power, the environmental impact would be minimal. However currently such processes are not viable, nevertheless the conversion of biomass to hydrogen, which although not carbon neutral, has a much lower contribution than fossil fuels, has a clear industrial potential especially when coupled to fuel cell applications. Hydrogen can be produced from oxygenated compounds and hydrocarbons, indeed the main source of hydrogen currently is steam reforming of methane.4 Among the various oxygenated compounds which can be converted to hydrogen, alcohols are very propitious candidates because they are easily decomposed in the presence of steam and, in principle, can generate a hydrogen-rich mixture suitable for fuel cells.7 Currently, extensive research is going on into the steam reforming of methanol both industrially and academically for hydrogen production. However, the main drawback of methanol is its high toxicity and that it is produced from non-renewable sources such as methane.8 The steam reforming of ethanol has attracted significant interest in the last few years because of its many advantages such as safe handling, easy transport, high solubility in water, low cost, low toxicity and sulphur free-composition.9–11 Moreover it is renewable from biomass such as sugar cane and sugar beets and produces 8–10 times more energy than consumed during processing.12
Hydrogen can be produced from ethanol in different ways such as oxidative steam reforming (eqn (1)), partial oxidation (eqn (2)) and steam reforming (eqn (3)). However steam reforming of ethanol gives the highest yield of hydrogen.13
| | | C2H5OH + 2H2O + 0.5O2 → 2CO2 + 5H2 ΔH298 = −69 kJ mol−1 | (1) |
| | | C2H5OH + 1.5O2 → 3H2 + 2CO2 ΔH298 = −552 kJ mol−1 | (2) |
| | | C2H5OH + 3H2O → 2CO2 + 6H2 ΔH298 = +174 kJ mol−1 | (3) |
There are also considerable energetic differences between these three routes.
Eqn (1) is exothermic and the heat liberated can be used to raise steam, hence reducing the overall energy requirements of the process.
Eqn (2) is exothermic and requires no steam raising, hence it can supply heat energy to other areas of the process/plant as well as hydrogen, whereas
eqn (3) is endothermic and significant heat is required both to sustain the reaction and to pre-heat the water to the required temperature. If excess water is used,
i.e. greater than stoichiometric, the energy demand increases and indeed this is a significant consideration in process economics.
4
The reaction pathway of steam reforming of ethanol is, however, complex and varies significantly with reaction parameters such as temperature, steam to ethanol ratio, catalyst, support and pressure. Some of the reactions that can occur during ethanol steam reforming (ESR) are detailed below:
| | | C2H5OH + H2O → 2CO + 4H2 ΔH298 = +256 kJ mol−1 | (4) |
| | | C2H5OH → CH3CHO + H2 ΔH298 = +69 kJ mol−1 | (5) |
| | | CH3CHO → CH4 + CO ΔH298 = −19 kJ mol−1 | (6) |
| | | CH3CHO + H2O → 3H2 + 2CO ΔH298 = +187 kJ mol−1 | (7) |
| | | 2CO → CO2 + C ΔH298 = −172 kJ mol−1 | (8) |
| | | CO + H2O → CO2 + H2 ΔH298 = −41 kJ mol−1 | (9) |
Ideally for hydrogen production, ethanol steam reforms and forms CO
2 and H
2 as shown in
eqn (3). However as can be seen in
eqn (4) ESR can also deliver an ideal
syn-gas for Fischer–Tropsch synthesis. Over catalysts which are less active and have no acidic sites, ethanol may dehydrogenate to form acetaldehyde and hydrogen
14 and the aldehyde may subsequently react to form acetic acid or acetone. Steam reforming of ethanol over catalysts having acidic sites may result in dehydration to form ethene and water (
eqn (10)). Ethene may then polymerize to form coke or undergo steam reforming to form hydrogen and carbon monoxide or carbon dioxide depending upon temperature and catalyst.
15| | | C2H5OH → C2H4 + H2O →ΔH298 = +45 kJ mol−1 | (10) |
| | | C2H4 → polymerize → coke + H2 | (11) |
| | | C2H4 + 2H2O → 2CO + 4H2 ΔH298 = +21 kJ mol−1 | (12) |
Steam reforming of ethanol is an endothermic reaction and temperature has a significant effect on the process.
7,16 The distribution of products significantly varies with temperature. At low temperature mostly C
2 products such as acetaldehyde and ethylene are produced. However at high temperature these products further decompose to form C
1 products or deposit on the catalyst forming coke. Wang
et al.16 investigated the steam reforming reaction at different temperatures for 8 h and reported that a small amount of ethanol conversion occurred up to 723 K, which decreased with reaction time. Performing the reaction at high temperature gave almost 100% conversion with no deactivation observed during the 8 h time on stream.
16 Liguras
et al.17 studied the steam reforming of ethanol over supported metal catalysts in the temperature range of 873–1073 K and found that with increased temperature the ethanol conversion significantly increased with high selectivity to CO, CO
2 and H
2.
17 Theoretical calculations for the steam reforming of ethanol showed that operating above 773 K significantly shifted the equilibrium position for carbon monoxide. They further explained that all other reactions favour an increase in the yield of hydrogen at higher temperature except the reverse water gas shift which has a negative effect on hydrogen yield.
17–19 Rabenstein
et al.20 performed theoretical calculation for the steam reforming of ethanol at different temperatures and different water to ethanol ratios. They concluded that more than 4 moles of hydrogen per mole ethanol can be obtained by using a water
:
ethanol ratio above 4 in the reaction temperature range between 823 K and 923 K. Using these conditions carbon monoxide can be reduced to 0.1 mole fraction in the effluent gas mixture.
20
Ruthenium has been shown to be one of the least active precious metals for ethanol steam reforming. Liguras et al.17 compared the catalytic performance of Rh, Ru, Pt, and Pd catalysts at 873–1123 K with metal loadings of 0–5 wt% and showed that Ru was inactive at low loadings but at 5 wt% loading Ru/Al2O3 could give a conversion of 95% at high temperatures. However Ramos et al.21 obtained high activity with a 2% Ru/CeO2/YSZ catalyst at temperatures >673 K, while Erdohelyi et al.22 obtained activity with 1 wt% Ru/alumina at 723 K.
The aim of this study is to investigate the steam reforming of ethanol over Ru/Al2O3 at different temperatures and high pressure. In this study the metal loading is 0.2%, which is more industrially relevant than the high loadings used in other studies and an industrially relevant pressure (20 barg) has been used to mirror a typical process load on the catalysts.
Experimental
The catalyst used throughout this project was a 0.2% Ru/Al2O3 prepared via incipient wetness impregnation. The alumina was determined by XRD analysis to be a mixture of γ- and δ-alumina phases. The metal salt used was Ru(NO)(NO3)3. After drying the catalysts were calcined at 723 K for 4 h. The BET surface area of the calcined catalysts was 104 ± 3 m2g−1, while the metal particle size and dispersion was determined from XRD, after reduction at 873 K for 2 h, to be 12.5 nm and ∼8.5% respectively. Catalysts were crushed and particle sizes between 600 to 425 μm were used for steam reforming reactions.
Ethanol steam reforming was performed in a continuous-flow, high-pressure, microreactor. Prior to reaction, catalysts (0.25 g) were reduced in situ at 873 K for 2 h using hydrogen gas at a flow rate of 50 ml min−1. The hydrogen was then purged from the system with argon and the temperature set to the desired reaction temperature. The pressure in the reactor was increased to 20 barg using argon. The water–ethanol solution was mixed in order to obtain steam to ethanol ratio of 5:1 in the gas phase. This ratio was chosen as one that is close to that found in fermentation ethanol. The ethanol–water solution was introduced to the reactor system through a vaporizer set at a temperature of 773 K. The gas flow rate of the steam/ethanol was set at 760 ml min−1, which was generated by pumping the liquids through a Gilson pump at a rate of 0.412 ml min−1. The argon gas flow rate was set at 10 ml min−1 achieving an overall GHSV of 50000 h−1. Once all the reaction parameters had been fixed, analysis was begun by flowing reactants from vaporizer to reactor. The eluant from the reactor tube in gaseous form entered a knockout pot where high boiling point products were collected and analysed by a Trace GC-2000 Series using a Zebron column and FID detector. The temperature of knockout pot was kept at 273 K. The gaseous products were analysed by an on-line Varian GC 3400 using a TCD detector and a carboxenTm1010 plot column. The reaction was performed at 773 K, 823 K and 873 K for 100 h time on stream.
Ethanol conversion was calculated as follows;
conversion = [(ethanol in − ethanol out)/ethanol in]
The yields for liquid products were calculated as follows;
yield = (moles of product X/moles of ethanol in)
The molar selectivity for the dry gas was calculated as follows;
selectivity of X = (moles of X/[Σ(moles of all gas phase products)]).
The amounts and nature of coke were determined by analysing post reaction catalyst samples using analytical techniques such as BET, powder XRD, Raman spectroscopy, SEM and TGA-DSC with evolved gas analysis.
BET surface areas and pore volume of pre- and post-reaction catalysts were measured using a Micromeritics Gemini III 2375 Surface Area Analyser. Prior to analysis, between 0.04–0.05g of catalyst were placed into a vial and purged under a flow of N2 (30 ml min−1) over night at 110 °C to remove moisture and any physisorbed gases from the catalyst sample. Powder X-ray diffraction patterns of pre and post reaction samples were obtained using a Siemens D 5000 X-ray Diffractometer (40 kV, 40 mA, monochromatic). The scanning range was 5° ≤ 2θ ≥ 85° with a scanning rate of 10 s per step and a step size of 0.02°. Raman spectra of post reaction catalysts were obtained with a Horiba Jobin Yvon LabRAM High Resolution spectrometer. A 532.17 nm line of a coherent Kimmon IK series He–Cd laser was used as the excitation source for the laser. Laser light was focused for 10 s using a 50× objective lens and grating of 600. The scattered light was collected in a backscattering configuration and was detected using nitrogen cooled charge-coupled detector. A scanning range of between 100 and 4100 cm−1 was applied. SEM images of the post reaction catalysts were obtained using a Philips XL30 Environmental SEM. The sample was irradiated with a beam of electrons, this was followed by changing magnification and focusing for increasing resolution of the catalyst surface. Temperature programmed oxidation (TPO) was carried out on post-reaction samples using a combined TGA/DSC SDT Q600 thermal Analyser connected to an ESS Mass Spectrometer for evolved gas analysis. Each sample was heated from room temperature to 1000 °C using a heating ramp of 5 deg min−1 under 2% O2/Ar at a flow rate of 100 ml min−1.
Results
Alumina
The alumina support was tested for ethanol steam reforming (ESR) in the absence of any metal. It was found to be active and converted all the ethanol. The main gaseous products at 873 K were hydrogen (44%), methane (28%), carbon dioxide (14%) and carbon monoxide (13%): only trace amounts of ethene and liquid products (∼1% acetone) were detected. Over the course of the 100 h on stream the alumina deactivated by <2%.
The used alumina was analysed by TGA using a 2% O2/Ar gas stream. There was a single, exothermic, weight-loss centred around 985 K. Mass spectrometry confirmed carbon dioxide as the sole product (Fig. 1): no significant amount of water was produced. The amount of carbon removed was 19.8% of the weight of the sample; this represents less than 0.03% of the carbon fed over the 100 h period. Raman spectroscopic analysis of the spent alumina revealed two bands at 1345 cm−1 and 1595 cm−1 (Fig. 2). These bands are well known to be representative of graphitic type carbon and have been designated the D and G band respectively.
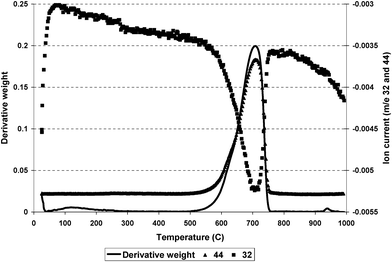 |
| | Fig. 1 Derivative weight loss and m/z 32 and 44 as a function of temperature for alumina subjected to a 2% O2/Ar flow. | |
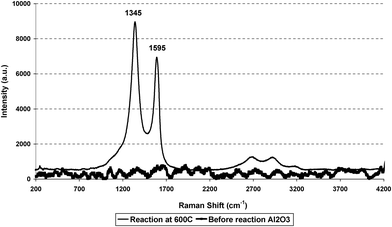 |
| | Fig. 2 Raman spectra of alumina, and alumina after use in ESR at 873 K. | |
Ru/Al2O3
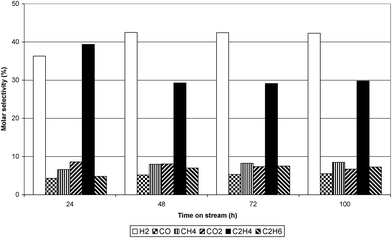
The conversion of ethanol at 773 K, 823 K and 873 K over the Ru/Al2O3 catalyst is shown in Fig. 3. The figure illustrates that in the initial one hour time on stream (TOS) ethanol conversion was almost 99% at all temperatures. However, after one hour TOS, deactivation of the catalyst took place and, at all reaction temperatures, the conversion of ethanol progressively decreased with time. The largest decrease in ethanol conversion was observed at 873 K while the decrease at 773 and 823 K was similar. Fig. 4–6 show selectivity to the various gas phase products. At 773 K an initial high selectivity to ethene was observed, which steadily decreased with time on stream. At 823 K ethene was the major product in the initial stages of experiment but was quickly supplanted by hydrogen. At 873 K hydrogen had the highest selectivity. As the temperature was increased the selectivity to methane and carbon monoxide increased significantly. The ratio of ethene:ethane suggested that ethene hydrogenation was inhibited given that the thermodynamic ratio should favour ethane. Table 1 reports the yield of liquid products over Ru/Al2O3 at 773 K, 823 K and 873 K. At 773 K the principal liquid products were acetaldehyde and diethyl ether. As the temperature was raised to 873 K acetaldehyde became the only significant liquid product.
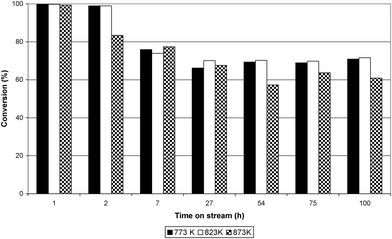 |
| | Fig. 3 Conversion (%) of ethanol over Ru/Al2O3 at 773 K, 823 K and 873 K. | |
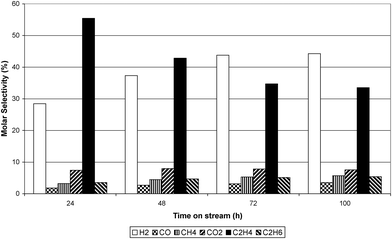 |
| | Fig. 4 Dry gas molar selectivity at 773 K. | |
 |
| | Fig. 5 Dry gas molar selectivity at 823 K. | |
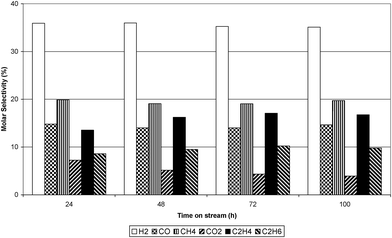 |
| | Fig. 6 Dry gas molar selectivity at 873 K. | |
Table 1 Yield of liquid products over Ru/Al2O3 as a function of temperature and time on stream
| Temp. (K) |
TOS (h) |
Yield of liquid products (%) |
| Acetald.a |
DEEa |
Acetone |
AAcida |
1,1-DEEa |
|
Acetald. = acetaldehyde, DEE = diethyl ether, AAcid = acetic acid, 1,1-DEE=1,1-diethoxy ethane.
|
| 773 |
24 |
5.8 |
2.1 |
2.7 |
0.8 |
0.0 |
| 48 |
4.1 |
3.8 |
2.2 |
1.0 |
0.3 |
| 72 |
3.7 |
3.4 |
1.7 |
1.1 |
0.3 |
| 100 |
3.1 |
3.5 |
1.2 |
0.8 |
0.2 |
| 823 |
24 |
4.4 |
0.8 |
1.8 |
0.7 |
0.2 |
| 48 |
3.7 |
2.3 |
1.2 |
0.7 |
0.2 |
| 72 |
3.7 |
2.3 |
1.0 |
0.8 |
0.2 |
| 100 |
4.0 |
2.3 |
0.9 |
0.9 |
0.2 |
| 873 |
24 |
4.9 |
1.1 |
1.1 |
0.4 |
0.2 |
| 48 |
10.3 |
1.4 |
1.4 |
1.5 |
0.3 |
| 72 |
11.1 |
1.5 |
1.2 |
1.4 |
0.4 |
| 100 |
10.2 |
1.5 |
1.0 |
1.3 |
0.4 |
Temperature programmed oxidation was carried out to identify the carbon species deposited on the catalyst after use in the steam reforming reaction. TGA, in a flow of 2% O2/Ar, of the post-reaction catalyst is shown in Fig. 7. The TGA profile shows that weight loss increased (389 to 444 mg g−1) when the temperature of the reaction was increased from 773 K to 873 K.
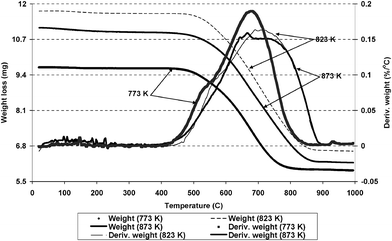 |
| | Fig. 7 Post reaction TGA in 2% O2/Ar of Ru/Al2O3 after use in ESR at 773 K, 823 K and 873 K. | |
At 873 K this weight loss represents 0.06% of the feed carbon over the 100 h on stream. The derivative weight profile shown in Fig. 8 illustrates that the main weight loss in the sample, after ESR at 773 K, occurred at 959 K, while there was also a small shoulder to the main weight loss event around 814 K. Similarly the derivative weight profile shows that in the samples subjected to ESR at 823 K and 873 K, the main loss was observed at around 973 K to 1003 K. The sole carbon containing species desorbed during the TGA was carbon dioxide. The carbon dioxide profile mirrored that of the derivative weight at all temperatures. Water was also produced. These results indicate that different types of coke were deposited on the catalyst surface at each temperature.
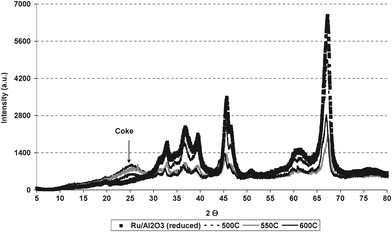 |
| | Fig. 8 Powder XRD patterns of reduced and post reaction Ru/Al2O3 after ESR at 773 K, 823 K and 873 K. | |
Spent samples were analysed by powder XRD to investigate coke composition and morphology of the catalyst after reaction at different temperatures as shown in Fig. 8. The powder XRD patterns illustrate that the particle size, calculated using Scherrer equation, of spent Ru/Al2O3 after use in ESR at 773 K is similar to the reduced catalyst (12.5 nm). However, the intensity of 823 K and 873 K reaction samples was decreased to small extent. The broad peak at a 2θ position of 26° suggests the presence of graphitic type coke over the catalyst.
Post-reaction samples were analysed by Raman spectroscopy, which gave two major bands at 1343 cm−1 and 1587 cm−1 as shown in Fig. 9. As the reaction temperature is increased the intensity and disorder of the graphitic species increased, as measured by the ratio in intensity of the G and D bands.
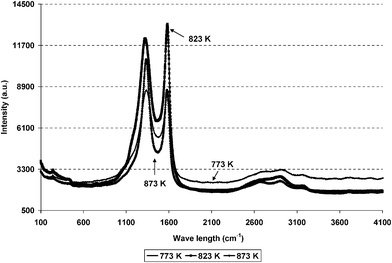 |
| | Fig. 9 Post reaction Raman spectra of reduced and post reaction of Ru/Al2O3 at 773 K, 823 K and 873 K. | |
SEM images (Fig. 10) show that on the 773 K post reaction sample, no filamentous type carbon was observed. This result suggests that at 773 K only amorphous coke is produced over Ru/Al2O3. However, as the temperature was increased from 773 K to 873 K the extent of filamentous carbon also increased. On the catalyst run at 873 K, besides filamentous coke, multi-wall carbon nanotubes appeared; these are only seen in the sample that had been used for ESR at 873 K.
 |
| | Fig. 10 SEM images of Ru/Al2O3 after ESR (a) at 773 K and (b) at 873 K. | |
BET analysis indicated that deposition of coke significantly reduced the surface area and average pore volume of the catalyst after use in the steam reforming reaction at all temperatures (Table 2). The decrease in the pore volume with increasing reaction temperature suggests that carbonaceous materials were mostly deposited in the pores of the catalyst. For further clarification, one of the samples was subjected to 2% O2/Ar at 873 K for 16 h and was then analysed by BET. The same BET surface area was obtained as the original catalyst after reduction. This result indicates that coke both blocks and fills the pores. Following regeneration at 873 K the sample was analysed by Raman spectroscopy and no significant change was observed in the graphitic bands. These results indicate that at least two types of coke; namely “soft” coke, which can be removed by oxygen below 873 K, and graphitic type carbon, which can only removed by oxygen at higher temperatures, exist on the catalyst surface.
Table 2 BET analysis, weight loss after TPO and Raman intensity ratios after ESR at 773 K, 823 K and 873 K
| Conditions (K) |
BET Surface Area(m2 g−1) |
Pore volume (cm3 g−1) |
Weight loss in TPO (%) |
(ID/IG) |
| Reduced@873 |
100 |
0.43 |
— |
— |
| 773 |
32 |
0.07 |
39 |
0.99 |
| 823 |
12 |
0.03 |
44 |
0.92 |
| 873 |
23 |
0.01 |
44 |
1.24 |
Discussion
Alumina
Although the alumina support was found to be active, it was not active for steam reforming. The product distribution reveals that the main reaction is ethanol decomposition followed by the water gas shift reaction:| | | 2C2H5OH → 2CO + 2CH4 + 2H2 ΔH298 = +99 kJ mol−1 | (13) |
| | | CO + H2O → CO2 + H2 ΔH298 = −41 kJ mol−1 | (9) |
overall,| | | 2C2H5OH + H2O → 3H2 + CO + 2CH4 + CO2 | (14) |
in agreement with the 3:2 ratio observed for H2:CH4 and the 1:1 ratio for (CO + CO2):CH4. The C:H:O ratio was calculated from the dry gas and compared with the theoretical ratio; the theoretical ratio was 1.3:4.7:1 while the calculated ratio was 1.3:4.8:1 confirming this reaction assessment. Ethanol decomposition and the WGS reaction have previously been observed over alumina in other studies.17,22,23 It is worthwhile noting that no significant deactivation of the alumina is observed and no extensive carbon is detected by SEM: however carbon was deposited. The carbon removed by TPO is unreactive requiring high temperatures (∼973 K) to initiate combustion. Raman spectroscopic analysis of the used support revealed the presence of graphitic species in keeping with the TPO. Clearly although these graphitic type species are present they do not significantly affect the activity of the support.
Ru/Alumina
It is important to note that the product distribution and deactivation is markedly different from that obtained with alumina in the absence of metal. For example no ethene was formed over the alumina in the absence of ruthenium, whereas in the presence of ruthenium, ethene is a major product. Therefore the alumina support in the absence of metal is not a good reference material as it is changed significantly by metal addition. The change of conversion of ethanol with time over the Ru/Al2O3 catalyst, shown in Fig. 2, reveals that there is a period of initial deactivation (<24 h) before steady state is established. This behaviour is in agreement with the literature,17 albeit using a higher metal loading, where it was reported that a 5% Ru/Al2O3 catalyst showed initial deactivation before achieving steady state. This loss in activity, which is due to carbon deposition, most likely from the polymerisation of ethene,24 will be discussed fully in a later section.
From the high yield of ethene at 773 K, especially in the early stages, it is clear that the main reaction was dehydration, forming ethene and water, rather than steam reforming. In the first hour on stream, no carbon monoxide is detected and it is possible to describe the reactions occurring as:
| | | 10C2H5OH → 10C2H4 + 10H2O ΔH298 = +450 kJ mol−1 | (10) |
| | | C2H5OH → CO + CH4 + H2 ΔH298 = +49 kJ mol−1 | (13) |
| | | CO + H2O → CO2 + H2 ΔH298 = −41 kJ mol−1 | (9) |
overall,
| 11C2H5OH + H2O → CH4 + CO2 + 2H2 + 10C2H4 + 10H2O |
However this gives an underestimate of the hydrogen produced. From the dry gas ratios the H
2:
CH
4 ratio is 7. This suggests that hydrogen is being produced from another source. As will be discussed later, this source is the deposited hydrocarbonaceous material. In principle ethene may hydrogenate to give ethane, polymerise to form coke or undergo steam reforming to form CO and H
2.
| | | C2H4 + H2 → C2H6 ΔH298 = −136 kJ mol−1 | (15) |
| | | C2H4 → Polymerise → coke + H2 | (11) |
| | | C2H4 + 2H2O → 2CO + 4H2 ΔH298 = +210 kJ mol−1 | (12) |
In the first few hours no ethane is produced and even later on in the run no significant amount of ethane is formed. This is slightly surprising given that the equilibrium position lies in favour of ethane (∼95% at 773 K) and that there is, in the gas phase at least, an excess of hydrogen. However it is clear that hydrogenation is not favoured, possibly due to the low level of hydrogen present on the surface. With time on stream the catalyst deactivates and the yield of ethene decreases however the yields of the other gaseous products, including ethane, increase. Hence during this period there is a shift from dehydration over the acid sites of the support to steam reforming, due to deactivation of the acid sites by carbon laydown from the ethene. Note that the support, in the absence of the metal, did not show any activity for ethanol dehydration suggesting that the acid sites were generated during the catalyst manufacturing procedure. Similar effects have been observed in other studies.
17,22,23
By 873 K there is a significant change in the dry gas product distribution. Ethene selectivity has decreased while the yields of ethane, methane and carbon monoxide have all dramatically increased. The C2H4:C2H6 ratio has changed from 15.6:1 at 773 K to 1.6:1 at 873 K in the first 24 h TOS and from 6.2:1 to 1.7:1 after 100h on stream. Therefore the extent of ethene hydrogenation has markedly increased. Assuming that ethane only comes from hydrogenation of ethene suggests that the pre-hydrogenation selectivity to ethene was >25%. This hydrogenation also reduces the hydrogen yield as a mole of hydrogen is used for every mole of ethane produced. Over the 773–873 K temperature range hydrogenation of ethene to ethane appears to be under kinetic control with C2H4:C2H6 ratios far removed from equilibrium. Therefore an activation energy was calculated from the data after 100 h TOS and a value of ∼46 kJ mol−1 obtained. This value is close to 35 kJ mol−1, the activation energy found around 300 K,25 which suggests that ethene hydrogenation is under kinetic control possibly due to the high level of carbon monoxide and/or the low surface concentration of hydrogen.
At 873 K the methane concentration has increased markedly. Methane may be formed from a number of different sources, the decomposition of acetaldehyde (whose yield also increased), hydrogenolysis of ethene or ethane, and ethanol decomposition. The (CO + CO2):CH4 ratio is close to 1:1 which would be indicative of decomposition of ethanol or acetaldehyde but the low hydrogen selectivity suggests that methane may be being formed via hydrogenolysis of ethene or ethane, which would remove hydrogen and along with ethene hydrogenation may explain the relatively low selectivity to hydrogen. The low carbon dioxide value is indicative of the shift in equilibrium position of the water gas shift reaction. Analysis suggests that approximately half the ethanol reacted is dehydrated to ethene, while the other half decomposes to carbon monoxide, methane and hydrogen, before the ethene and carbon monoxide further react to give ethane and carbon dioxide respectively. Little steam is converted to hydrogen and the productivity gives an ethanol fed to hydrogen produced ratio of 1.2:1.
| | | 6C2H5OH → 6C2H4 + 6H2O ΔH298 = +270 kJ mol−1 | (10) |
| | | 2C2H4 + 2H2 → 2C2H6 ΔH298 = −272 kJ mol−1 | (15) |
| | | 5C2H5OH → 5CO + 5CH4 + 5H2 ΔH298 = +250 kJ mol−1 | (13) |
| | | CO + H2O → CO2 + H2 ΔH298 = −41 kJ mol−1 | (9) |
overall,
| 11C2H5OH → 5CH4 + CO2 + 4CO + 4H2 + 4C2H4 + 2C2H6 + 5H2O |
Once again this underestimates the hydrogen evolution observed. From the dry gas ratios the H
2:
CH
4 ratio is 1.8 whereas the above analysis gives a ratio of 0.8, indicating the liberation of hydrogen from the surface deposit.
The formation of acetaldehyde and other liquid products indicate that not only is ethanol dehydrated over the Ru/Al2O3 catalyst but it also undergoes dehydrogenation. The dehydrogenation reaction can take place over the support and metal.23,26 The presence of diethylether formed by acid catalysed reaction of ethanol (eqn (16)) confirms the presence of acidic sites on the alumina. Ethyl acetate can similarly be formed from ethanol and acetic acid (eqn (17)), while 1,1-diethoxy ethane is the acetal formed by acid catalysed reaction between ethanol and acetaldehyde (eqn (18)). The formation of acetic acid is slightly surprising but can be viewed as an intermediate in the oxidation of ethanol to carbon dioxide.
| | | 2C2H5OH → C2H5OC2H5 + CO + H2O ΔH298 = +48 kJ mol−1 | (16) |
| | | C2H5OH + CH3COOH → CH3COOC2H5 + H2O ΔH298 = −56 kJ mol−1 | (17) |
| | | 2C2H5OH + CH3CHO → CH3CH(OC2H5)2 + H2O ΔH298 = −97 kJ mol−1 | (18) |
The extent of carbon deposition is related to temperature, with this difference reflected in the weight loss observed by TGA. Analysis of the reactor eluant suggests that most of the deposition occurs in the early stages of the reaction (<30 h). At 773 K there is no visible evidence, from the SEM analysis, of carbon filaments or other defined carbon structures, however the TGA/TPO and Raman spectra clearly show evidence of carbon deposition and the BET surface area is also reduced. The Raman spectrum and the high temperature required to combust the deposit indicate that the deposit is graphitic and will have a low H:C ratio, hence we may expect that the deposit will liberate hydrogen with time as it converts from a species with a high H:C ratio (e.g. ethene 2:1, ethanol 3:1, methane 4:1). The temperature limited TPO reveals that it is the “soft” coke that blocks the pores, while the graphitic material has little effect on surface area.
Can et al.27 studied the steam reforming of ethanol over Rh/Al2O3 at 673 K and found that after 8 h four different types of coke were formed on the catalyst surface. They identified four carbon combustion events in the TPO at (I) 583 K, (II) 653 K, (III) 793 K and (IV) 968 K and assigned type I to coke deposited on the metal, type II to coke deposited around the metal-support interface, type III to carbon deposited on the support, while type IV was assigned to a graphite phase generated by the thermal ethanol decomposition of ethanol.27 Our results confirm their assignment of the type IV material. In the absence of metal, after use in ESR, alumina gives a TPO peak at 985 K, while the Raman spectrum confirms the presence of graphitic species, and analysis of the reaction products confirmed that only ethanol decomposition was taking place. However we see no evidence of combustion at 583 K and 653 K over Ru/alumina. It may be that these species are only present at low temperature (673 K cf. 773–873 K) or that the TOS makes a difference (8 h cf. 100h). Nevertheless if they are associated with the metal and the metal/support interface we would have expected to have seen them. A combustion event was observed at ∼813 K, which would agree with the type III combustion, however if, as Can et al.27 suggest, this material is on the support then the question arises as to why it is not seen when no metal is present. This would suggest that the type III carbonaceous species is formed on the metal and spillsover from the metal to the support.
Following ESR at 873 K, analysis by SEM revealed the presence of filamentous/whisker carbon and carbon nanotubes (CNTs). The formation of CNTs over metals other than nickel and iron is not well documented but a recent paper28 has shown that CNT materials can be formed over Pt from ethene. Other reports29 have shown that the ability to form CNT materials from hydrocarbon gases over metals is more widespread than simply iron and nickel. Nevertheless we believe that this is one of the first reports of CNT formation catalysed by ruthenium. Ramos et al.21 observed carbon filaments over a Ru/CeO2/YSZ after ESR at 823 K but to a much lesser extent. In their system the re-oxidation of the reduced ceria masks the combustion of carbon in the TPO and hence we are unable to make a comparison. Given the significant ethene production it is likely that most of the carbon deposition comes from this source but at high temperature (873 K) cracking of CH4 and CO can also play a role in coke deposition.30 Although CNTs have not been reported from ESR reactions, ethanol has been used as a feedstream.31 Hart and co-workers31 used ethanol as a modifier to an ethene/hydrogen feed and found that the presence of ethanol increased the rate of carbon deposition. They also observed that the steam formed from the ethanol reacted with amorphous carbon giving a higher purity CNT.31 The Raman results suggest that with an increase of the reaction temperature there is an increase in the disorder of the graphitic type carbon giving an ID/IG ratio of 1.24 (Table 2). This is in contrast to the CNT studies where typically the ID/IG ratio is 0.7 at 873 K.32 Therefore this increase in disorder may be due to an increased coke contribution from CO and/or CH4 by the Boudouard reaction and cracking to give amorphous carbon species. However the TPO profiles obtained by Wang et al.32 did not match those found in our study, confirming that the carbon deposit under steam reforming conditions is far more complex than just CNTs. Nevertheless the formation of these species will result in hydrogen production as initially adsorbed species convert to CNTs and other dehydrogenated graphitic species.
Conclusions
In this study we have shown that a catalyst with 0.2 wt% loading of ruthenium is active for ESR between 773 K and 873 K. The conversion of ethanol, especially at 873 K, can be described in terms of ethanol decomposition, ethanol dehydration and the WGS reaction resulting in a hydrogen yield per mole of ethanol consumed of 0.8. Hydrogen was the major product in the steady state but ethene was a significant product especially at 773 and 823 K. The hydrogenation of ethene to ethane is significantly inhibited over Ru/Al2O3 sufficient for a sensible activation energy to be determined.
The liquid products detected reveal the complexity of the reaction sequence with products formed requiring a cascade of reactions (e.g. acetone). They also reveal that most of the non-decomposition reactions are acid or base catalysed and do not, in themselves, require a metal to be present.
Catalyst deactivation was shown to be due to carbon deposition. At all temperatures the TPO profile shows an event at high temperatures indicative of graphitic species, this is supported by the Raman spectra. At 873 K CNTs are formed, which appears to be one of the first example of CNT formation over ruthenium. Although CNTs are present on the catalyst surface it is likely that they have little effect on catalyst activity, rather it is the other carbonaceous species that cause the reduction in activity. The existence of such species can be seen from the limited temperature TPO, the Raman spectra having a different IG/ID ratio from those in the literature representative of CNTs and the full TPO is different from that found in pure CNT systems. It is likely that ethene is the major coke precursor with contributions from carbon monoxide and methane. However although filamentous carbon/CNTs may not directly affect activity they are a major issue in industrial application. In steam reforming of hydrocarbons over nickel catalysts it is important to avoid the formation of CNTs or whisker carbon.33 The CNT/whisker is strong and, although the property is much desired in nanotechnology, it has the potential to cause significant damage in a steam reformer.34 CNTs can easily fracture a catalyst pellet when they come into contact with a pore wall and in a reformer tube broken catalyst pellets and carbon can cause maldistribution of the feed gas leading to overheating of tubes and, in the limit, result in tube failure. Therefore the presence of CNT/whisker species in steam reforming must be avoided. The presence of these species in ESR over Ru/alumina is an issue that must be resolved before large scale application could be envisaged.
References
- O. Gorke, P. Pfeifer and K. Schubert, Appl. Catal., A, 2009, 360, 232–241 CrossRef.
- M. Ni, D. Y. C. Leung, M. K. H. Leung and K. Sumathy, Fuel Process. Technol., 2006, 87, 461–472 CrossRef CAS.
- S. Zinoviev, F. Muller-Langer, P. Das, N. Bertero, P. Fornasiero, M. Kaltschmitt, G. Centi and S. Miertus, ChemSusChem, 2010, 3, 1106–1133 CrossRef CAS.
- J. R. Rostrup-Nielsen and T. Rostrup-Nielsen, CATTECH, 2002, 6, 150–159 CrossRef CAS.
- T. Nishiguchi, T. Matsumoto, H. Kanai, K. Utani, Y. Matsumura, W.-J. Shen and S. Imamura, Appl. Catal., A, 2005, 279, 273–277 CrossRef CAS.
- A. Bshish, Z. Yakoob, B. Narayanan, R. Ramakrishnan and A. Ebshish, Chem. Pap., 2010, 65, 251–266 CrossRef.
- M. A. Goula, S. K. Kontou and P. E. Tsiakaras, Appl. Catal., B, 2004, 49, 135–144 CrossRef CAS.
- P. Biswas and D. Kunzru, Chem. Eng. J., 2008, 136, 41–49 CrossRef CAS.
- J. Llorca, N. Homs, J. Sales and P. R. de la Piscina, J. Catal., 2002, 209, 306–317 CrossRef CAS.
- R. C. Cerritos, R. F. Ramirez, A. F. A. Alvarado, J. M. M. Rosales, T. V. Garcia and I. R. G. Esquivel, Ind. Eng. Chem. Res., 2011, 50, 2576–2584 CrossRef.
- H. Devianto, J. Han, S. P. Yoon, S. W. Nam, T.-H. Lim, I.-H. Oh, S.-A. Hong and H.-I. Lee, Int. J. Hydrogen Energy, 2011, 36, 10346–10354 CrossRef CAS.
- A. C. Furtado, C. G. Alonso, M. P. Cantão and N. R. C. Fernandes-Machado, Int. J. Hydrogen Energy, 2009, 34, 7189–7196 CrossRef CAS.
- O. Akdim, W. Cai, V. Fierro, H. Provendier, A. van Veen, W. Shen and C. Mirodatos, Top. Catal., 2008, 51, 22–38 CrossRef CAS.
- M. Ni, D. Y. C. Leung and M. K. H. Leung, Int. J. Hydrogen Energy, 2007, 32, 3238–3247 CrossRef CAS.
- S. Cavallaro, Energy Fuels, 2000, 14, 1195–1199 CrossRef CAS.
- H. Wang, Y. Liu, L. Wang and Y. N. Qin, Chem. Eng. J., 2008, 145, 25–31 CrossRef CAS.
- D. K. Liguras, D. I. Kondarides and X. E. Verykios, Appl. Catal., B, 2003, 43, 345–354 CrossRef CAS.
- I. Fishtik, A. Alexander, R. Datta and D. Geana, Int. J. Hydrogen Energy, 2000, 25, 31–45 CrossRef CAS.
- A. Lima da Silva, C. d. F. Malfatti and I. L. Müller, Int. J. Hydrogen Energy, 2009, 34, 4321–4330 CrossRef CAS.
- G. Rabenstein and V. Hacker, J. Power Sources, 2008, 185, 1293–1304 CrossRef CAS.
- I. A. C. Ramos, T. Montini, B. Lorenzut, H. Troiani, F. C. Gennari, M. Graziani and P. Fornasiero, Catal. Today, 2012, 180, 96–104 CrossRef CAS.
- M. Dömök, M. Tóth, J. Raskó and A. Erdőhelyi, Appl. Catal., B, 2007, 69, 262–272 CrossRef.
- F. Aupretre, C. Descorme, D. Duprez, D. Casanave and D. Uzio, J. Catal., 2005, 233, 464–477 CrossRef CAS.
- F. Aupretre, C. Descorme and D. Duprez, Catal. Commun., 2002, 3, 263–267 CrossRef CAS.
-
G. C. Bond, Metal-catalysed reactions of hydrocarbons (fundamental and applied catalysis), Springer, New York, 2005 Search PubMed.
- M. Dobrovolszky, P. Tétényi and Z. Paál, J. Catal., 1982, 74, 31–43 CrossRef CAS.
- F. Can, A. Le-Valant, N. Bion, F. Epron and D. Duprez, J. Phys. Chem. C, 2008, 112, 14145 CAS.
- S. Takenaka, T. Iguchi, E. Tanabe, H. Matsune and M. Kishida, Carbon, 2009, 47, 1251–1257 CrossRef CAS.
- D. Yuan, L. Ding, H. Chu, Y. Feng, T. P. McNicholas and J. Liu, Nano Lett., 2008, 8, 2576–2579 CrossRef CAS.
-
E. Opara, S.D. Jackson, unpublished results.
- Y. Zhang, J. M. Gregoire, R. B. van Dover and A. J. Hart, J. Phys. Chem. C, 2010, 114, 6389–6395 CAS.
- G. Wang, H. Wang, W. Li, Z. Ren, J. Bai and J. Bai, Fuel Process. Technol., 2011, 92, 531–537 CrossRef CAS.
- S. Helveg, J. Sehested and J. R. Rostrup-Nielsen, Catal. Today, 2011, 178, 42–46 CrossRef CAS.
-
J. R. Rostrup-Nielsen and L. J. Christiansen, Concepts in Syngas Manufacture, Imperial College Press, London, 2011 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here.