Kinetic aspects and deactivation behaviour of chromia-based catalysts in hydrogen chloride oxidation
Amol P.
Amrute
,
Cecilia
Mondelli
and
Javier
Pérez-Ramírez
*
Institute for Chemical and Bioengineering, Department of Chemistry and Applied Biosciences, ETH Zurich, Wolfgang-Pauli-Strasse 10, CH-8093 Zurich, Switzerland. E-mail: jpr@chem.ethz.ch; Fax: +41 44 633 1405
First published on 1st June 2012
Abstract
The gas-phase HCl oxidation was studied over bulk and supported Cr2O3-based catalysts by means of kinetic experiments in a fixed-bed reactor at ambient pressure and variable temperature, inlet O2/HCl ratio, and contact time. Cr2O3 exhibited high activity for Cl2 production. X-ray diffraction of the used catalysts detected no phase change. Temperature-programmed reduction with hydrogen and X-ray photoelectron spectroscopy showed that the surface of the fresh catalyst contains chromium species in oxidation state (III) as well as higher oxidation states (V and VI) while that of the used sample features the reduction of Cr5+/Cr6+ and comprises a little amount of chlorine. Coupling the catalytic and characterisation data over Cr2O3 with activity tests over CrO3, a redox cycle is proposed in which chromium species shift between Cr3+ and Cr5+ + Cr6+. The positive dependence of the HCl conversion on the inlet O2 concentration suggests that catalyst re-oxidation is rate limiting. SiO2 was identified as a better carrier for Cr2O3 than Al2O3 and TiO2-anatase, as it favours the formation of Cr2O3 nanoparticles rather than (unstable) isolated chromate species. However, all the supported catalysts suffered from severe deactivation due to substantial chromium loss. The deactivation mechanism is assigned to the in situ formation of the highly volatile CrO2Cl2 from Cr6+ species and CrO2(OH)2 from both Cr3+ and Cr6+ species. The deactivation rate can be reduced, though not suppressed, by applying a high O2 excess in the feed mixture, thus indicating that the deactivation route via the oxychloride might be predominant. The features observed represent critical reasons justifying the restricted industrial implementation of chromium-based catalysts for HCl oxidation.
1. Introduction
The catalysed oxidation of HCl to Cl2 has re-gained increasing interest as an energy-efficient route to recover chlorine from HCl-containing streams in the chemical industry, especially in polyurethane and polycarbonate production.1 Since the introduction of the original CuCl2/pumice catalyst by Deacon in 1868,2 many copper-based systems have been reported in the literature for operation in fixed- and fluidised-bed reactors.3–5 However, none of them were realised in a long-term commercial process as a consequence of the fast catalyst deactivation due to copper loss, operational problems such as particle coagulation, and severe corrosion issues in the plants. In 1980, Mitsui Chemicals established the MT-Chlor process using a Cr2O3/SiO2 catalyst in a fluidised-bed reactor at 623–703 K.6,7 The stability of the latter catalyst was improved with respect to the CuCl2–KCl/SiO2 catalyst of the Shell-Chlor process.3 This has been related to the fact that chromium(III) oxide operates via a redox cycle, without melting and without involving the chloride-oxide reaction cycle characteristic of copper-based catalysts.8,9 Nonetheless, the implementation of Mitsui's process is limited to one medium-sized plant of 60 kton Cl2 per year.8 In contrast, recently developed ruthenium-based catalysts are being installed in several large-scale Cl2 production facilities.8 RuO2-based materials exhibit a very high activity at low temperatures (550–650 K) and their lifetimes exceed several thousand hours.8,10–12 Thus, these catalytic systems appear to satisfy a long-standing industrial need to count on development of complementary technologies for the manufacture of Cl2 by electrolysis.The practical interest in RuO2-based catalysts has triggered investigations leading to in-depth knowledge on the catalyst structure, the chlorination behaviour, and the reaction mechanism and kinetics.13–19 Keeping in mind that the ruthenium cost associated with a world-scale HCl oxidation plant equipped with a ruthenium-based material easily amounts to several million euros,1 recent studies have attempted the identification of cheaper alternatives. The use of cuprous delafossite (CuAlO2), displaying outstanding stability in comparison with conventional copper-based catalysts,20 and the promising performance of CeO221 are representative examples. Astonishingly, the number of academic studies embracing HCl oxidation over chromium-based materials is limited,22,23 which hampers a proper understanding of this interesting catalytic system. Short-term isothermal tests have shown that Cr2O3 is more active than other metal oxides such as CuO and CeO2, being only surpassed by RuO2.23 One of the original patents by Mitsui7 states that fresh catalyst particles should be fed to the fluidised-bed reactor either continuously or intermittently to replenish the evaporated portion of the chromium while continuing the reaction. Therefore, chromium loss and the associated environmental concerns might be critical factors for the limited industrialisation.
Herein, we have conducted a systematic investigation of the gas-phase oxidation of HCl over Cr2O3-based catalysts in bulk and supported forms using a continuous-flow fixed-bed reactor operated at ambient pressure and variable temperature, feed O2/HCl ratio, and contact time. The composition, structure, porosity, and chromium oxidation state of the catalysts prior to and after reaction were assessed in order to gain insights into the nature of the active species and the deactivation mechanism.
2. Experimental
2.1. Catalysts and characterisation techniques
Cr2O3 (Aldrich, nanopowder, 99%), SiO2 (Fluka, Cab-O-Sil), γ-Al2O3 (Alfa Aesar), and TiO2-anatase (Aldrich, nanopowder, 99.7%) were calcined in static air at 773 K (10 K min−1) for 5 h prior to their use. CrO3 (Aldrich, 99.99%) was used as received. Supported chromium catalysts with a nominal Cr loading of 14 wt% were prepared by incipient wetness of the dried carriers with an aqueous solution of Cr(NO3)3·9H2O (Sigma-Aldrich, 99%), followed by drying at 338 K for 12 h, and static-air calcination at 773 K (10 K min−1) for 5 h. The value of 14 wt% was selected on the basis of the optimal range (13–20 wt%) indicated in the patent literature.6Powder X-ray diffraction (XRD) was measured on a PANanalytical X’Pert PRO-MPD diffractometer. Data were recorded in the 10–70° 2θ range with an angular step size of 0.017° and a counting time of 0.26 s per step. N2 sorption was measured at 77 K in a Quantachrome Quadrasorb-SI gas adsorption analyser. The samples were degassed in vacuum at 473 K for 12 h prior to the measurement. Temperature-programmed reduction with hydrogen (H2-TPR) was measured in a Thermo TPDRO 1100 unit equipped with a thermal conductivity detector. The samples were loaded in a quartz micro-reactor (11 mm id), pre-treated in He (20 cm3 STP min−1) at 473 K for 30 min, and cooled to 323 K in He. The analysis was carried out in 5 vol% H2/N2 (20 cm3 STP min−1), ramping the temperature from 323 to 1173 K at 10 K min−1. The chromium content was determined by inductively coupled plasma-optical emission spectroscopy (ICP-OES) in a Horiba Jobin Yvon Ultima 2 instrument after dissolution of the samples in a HF/H2SO4 solution. X-ray photoelectron spectroscopy (XPS) was performed on a VG-Microtech Multilab 3000 spectrometer featuring a hemispheric electron analyser with 9 channeltrons (pass energy 50 eV) and non-monochromatised Mg Kα radiation at 1253.6 eV as the X-ray source. Samples were transferred to the spectrometer chamber under regular ambient exposure. The binding energy scale was referenced to the C 1s level of the carbon overlayer at 284.6 eV. The spectrum of fresh Cr2O3 was deconvoluted by applying fixed binding energy values for Cr3+ and Cr6+, retrieved from NIST,24–26 and allowing the full width at half maximum to relax to achieve the best possible fit (R2 = 0.996). The same procedure was followed for the decomposition of the spectrum of used Cr2O3 by initially applying a shift of 2 eV to the binding energies of Cr3+ and Cr6+, on the basis of the overall shift of the Cr 2p features with respect to those of the fresh sample. As the fit was not satisfactory (R2 = 0.971), the binding energies were refined. The best result (R2 = 0.991) was found for shifts equal to 2.6 and 1.9 eV for Cr3+ and Cr6+, respectively. The point of zero charge (PZC) of Al2O3 and SiO2 was determined by measuring the zeta potential as a function of pH at different ionic strengths in a Zetasizer Nano ZS (Malvern Instruments). The carriers (0.1 wt%) were dispersed in aqueous solutions of pH 2.0 (Al2O3) or 11.2 (SiO2). The suspensions were placed in a cell and titrated by adding 0.01–0.1 M NaOH (Al2O3) or 0.01–0.1 M HCl (SiO2). The changes in zeta potential were recorded as a function of pH.
2.2. Catalytic tests
The gas-phase oxidation of hydrogen chloride was studied at ambient pressure in a setup described elsewhere,27 which is equipped with (i) mass-flow controllers to feed HCl (Messer, purity 2.8, anhydrous), O2 (Pan Gas, purity 5.0), and N2 (Pan Gas, purity 5.0), (ii) a home-made electrically-heated oven hosting a 8 mm id quartz micro-reactor, and (iii) a Mettler Toledo G20 Compact Titrator for Cl2 analysis. The catalysts (particle size = 0.4–0.6 mm) were loaded in the tubular reactor and pre-treated in N2 at 688 K for 30 min. Thereafter, experiments at variable bed temperatures (Tbed = 560–680 K), inlet O2/HCl ratios (0–7), and contact times (τ = 0.1–0.3 s) were carried out.In the catalytic tests, the inlet HCl concentration was fixed at 10 vol%. The temperature dependence was measured using a catalyst weight (W) of 0.5 g (0.25 g for supported catalysts), a O2/HCl ratio = 2, and a total volumetric flow (FT) of 166 cm3 STP min−1. The O2/HCl dependence was measured by increasing the O2 content in the inlet mixture from 5 to 70 vol% with N2 as balance gas, applying Tbed = 688 K (678 K for supported catalysts), W = 0.5 g (0.25 g for supported catalysts), and FT = 166 cm3 STP min−1. Data were collected after 1 h on stream under each condition. The contact time dependence was studied at variable W = 0.5–1.5 g and FT = 166–333 cm3 STP min−1, at Tbed = 688 K and O2/HCl = 2. In this case, measurements were made at 1, 2, and 3 h on stream and the data averaged. The space time, defined as the ratio of the catalyst weight and the inlet molar flow of HCl (W/ṅ0(HCl)), was in the range of 11.2–33.3 g h mol−1. The Weisz–Prater criterion was fulfilled in all our catalytic tests, indicating the absence of intra-particle diffusion limitations. Stability tests (up to 40 h on stream) were conducted over the supported Cr2O3 catalysts at 678 K and O2/HCl = 0.5–7. Used samples were collected for post-mortem characterisation after rapidly cooling down the reactor to room temperature in N2 flow. The percentage of HCl conversion was determined as XHCl = (2 × mole Cl2 at the reactor outlet/mole HCl at the reactor inlet) × 100. The experimental error of the measurements is ±3%. Chromium losses in stability tests involving bulk Cr2O3 were determined using a guard bed of H-ZSM-5 (Zeolyst International, CBV 8014, 0.5 g, particle size = 0.4–0.6 mm) located in the cold zone of the reactor outlet (373 K).
3. Results and discussion
3.1. Bulk Cr2O3
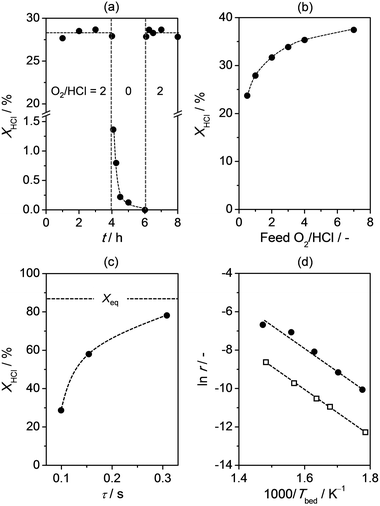 | ||
| Fig. 1 HCl conversion versus (a) time-on-stream, (b) feed O2/HCl ratio, and (c) contact time. (d) Arrhenius plot showing the logarithm of the rate of Cl2 production as a function of the reciprocal temperature. Bulk Cr2O3 is represented by solid circles and Cr2O3/SiO2 by open squares. The equilibrium HCl conversion is depicted as a dashed line (c). Conditions are detailed in Section 2.2. | ||
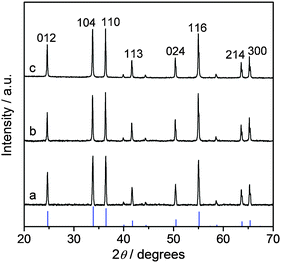 | ||
| Fig. 2 XRD patterns of bulk Cr2O3: (a) fresh, (b) used in O2/HCl = 2 at 688 K for 4 h, and (c) used in O2/HCl = 0 at 688 K for 2 h. The pattern of Cr2O3 (JCPDS 70-3766) is shown by the vertical blue lines. | ||
The H2-TPR profile of fresh Cr2O3 displays two main reduction peaks (1 and 2, Fig. 3a). Due to variations in the experimental conditions applied in our analysis (2–16 times lower concentration of H2 and 1.5 times slower ramping rate), these are observed at higher temperatures (500–650 and 650–850 K) with respect to literature data.28,29 The less intense low-temperature feature seems to be composed of a peak and a shoulder (1 and 1′, visualised by deconvolution as blue curves), which are respectively attributed to the reduction of Cr6+ to Cr3+28 and of Cr5+ to Cr3+. The latter assignment was substantiated by H2-TPR and XRD data for CrO3 after use in HCl oxidation (Section 3.1.3). As no reflections or bumps specific to Cr6+ or Cr5+ phases were detected by XRD, these species seem to be present at the near-surface level only. Peak 2 appears as the superimposition of a main component, related to the reduction of Cr3+ to Cr2+,28,29 and two shoulders (2′ and 2′′, visualised by deconvolution as blue curves) that could refer to variable degrees of crystallisation of Cr2O3.29 After use at O2/HCl = 2 (b), peak 1′ became moderately more intense, whereas peak 1 almost vanished. These findings indicate that HCl oxidation is accompanied by an increase of Cr5+ species and depletion of Cr6+ species, which might occur either via volatilisation or reduction (vide infra). The signals were fully depleted after treatment in HCl-only (c). In the absence of O2, HCl is able to consume all Cr6+ and Cr5+ species transforming them into Cr3+ species or, vice versa, Cr6+ and Cr5+ species oxidise HCl to Cl2. Chlorine production was indeed observed under these conditions and eventually stopped when all chromium species in higher oxidation states had reacted (Fig. 1a). The subsequent exposure to a feed with O2/HCl = 2 restored feature 1′ to a minor extent (d), demonstrating that gas-phase O2 is able to in situ re-oxidise Cr3+ species. In profiles (b–d) peak 2 is shifted to lower temperatures, indicating that any treatment renders Cr2O3 somewhat more reducible.29
 | ||
| Fig. 3 H2-TPR profiles of bulk Cr2O3: (a) fresh, (b) used in O2/HCl = 2 at 688 K for 4 h, (c) used in O2/HCl = 0 at 688 K for 2 h, and (d) used in O2/HCl = 0 at 688 K for 2 h and then in O2/HCl = 2 at the same temperature for 2 h. | ||
XPS was applied to further characterise the surface of the samples (Fig. 4). The band structure of the Cr 2p XPS spectrum for the fresh material (Fig. 4a), composed of the Cr 2p3/2 and Cr 2p1/2 peaks, is that characteristic of bulk Cr2O3.28 Both features can be fitted by contributions of Cr3+, Cr5+, and Cr6+. Nevertheless, due to the equivocal distinction between the binding energies of Cr6+ and Cr5+ (Table 1),30 a combined deconvolution line is shown for both. In order to derive information about the relative amounts of the different chromium species, the Cr 2p3/2 signals were considered.31 Accordingly, Cr5+ + Cr6+ species were significantly more abundant than Cr3+ species in fresh Cr2O3 (Fig. 4a). Upon use in HCl oxidation, both Cr 2p3/2 and Cr 2p1/2 peaks were shifted by 2 eV to higher binding energy (Fig. 4b), pointing to the presence of chlorine. Surrounding atoms with high electronegativity are indeed reported to cause an increase in binding energy.30 The noisy signal detected at 201.3 eV in the Cl 2p region of the XPS spectrum confirms that the surface of the used catalyst contains a little amount of chlorine (Fig. 4, inset). It is difficult to speculate on the type(s) of chlorine-containing species present. The signal of Cr3+ appears at 1.3 eV higher binding energy in the case of CrCl3 with respect to Cr2O3 and larger shifts are observed when additional ligands such as en or NH3 are present (Table 1). Shifts to higher binding energy are expected also in the case of substitution of O atoms bound to Cr5+ or Cr6+ by Cl. Thus, species likely present on the used sample could be surface (oxy/hydroxy)chlorides of chromium(III/V/VI), possibly solvated by water, but adsorbed hydrochloric acid and molecular chlorine cannot be excluded. As the individual shifts for Cr3+ and Cr5+ + Cr6+ are actually different and equal to 2.6 eV and 1.9 eV, respectively, chlorine might preferably bind to Cr3+ rather than Cr5+/Cr6+ ions, or Cr sites with different oxidation state could stabilize chlorine-containing compounds of different chemical nature. Finally, the Cr5+ + Cr6+ and Cr3+ contributions to the Cr 2p3/2 feature became almost equivalent. The depletion of species in higher oxidation states is in line with the H2-TPR results (Fig. 3, peaks 1 and 1′ in profiles a,b).
 | ||
| Fig. 4 Cr 2p core level XPS spectra of bulk Cr2O3: (a) fresh and (b) used in O2/HCl = 2 at 688 K for 4 h. The inset shows the Cl 2p spectrum of the used sample. | ||
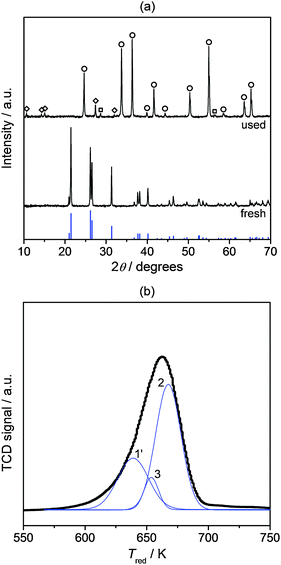 | ||
| Fig. 5 (a) XRD patterns of bulk CrO3 prior to and after use in O2/HCl = 2 at 688 K for 2 h. The pattern of CrO3 (JCPDS 32-0285) is shown by the vertical blue lines, while other phases are indicated by symbols: (○) Cr2O3, JCPDS 70-3766, (◇) Cr2O5, JCPDS 07-0247, and (□) CrO2, JCPDS 84-1821. (b) H2-TPR profile of used CrO3. | ||
XPS and H2-TPR analyses indicated the presence of Cr6+ and Cr5+ species in fresh Cr2O3. Production of Cl2, although in small amounts, in the absence of gas-phase O2 over this catalyst was observed up to 2 h on stream (Fig. 1a). After this time no HCl was further converted and H2-TPR analysis of the used sample evidenced the complete depletion of Cr6+ and Cr5+ species (Fig. 3c). Furthermore, CrO3 was active for HCl oxidation. These lines of evidence prove that Cr3+ species are per se not able to oxidise HCl, while species in higher oxidation states are. The fact that CrO3 reaches a lower HCl conversion level than Cr2O3, in spite of the exclusive presence of Cr6+ species, is likely due to its rapid volatilisation and decomposition. The instability of chromium(V) and (VI) compounds both at the calcination and reaction temperature, the absence of diffraction lines or bumps specific to bulk phases with chromium in higher oxidation state, and the detection of Cr5+ and Cr6+ species by XPS suggest that the latter are exclusively present at the surface of Cr2O3. Thus, HCl oxidation seems to occur at the catalyst surface. Oxidised chromium species can be generated during reaction in the presence of gas-phase O2. This is supported by (i) the disappearance of the reduction peak of Cr6+/Cr5+ species in the H2-TPR profile of the sample exposed to HCl-only (Fig. 3c) along with (ii) the immediate increase in HCl conversion to its original level (ca. 30%) when feeding a mixture with O2/HCl = 2 after the treatment at O2/HCl = 0 (Fig. 1a) and the re-appearance of the reduction peak of Cr5+ species in the H2-TPR profile of the sample after this test (Fig. 3d). The literature reports that surface chromyl species (Cr![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) can form via dissociative adsorption of O2 on coordinatively-unsaturated Cr3+ cations, i.e. transforming Cr3+ into Cr5+ and completing its coordination shell.35–37 The generation of chromyl terminations with Cr in (VI) oxidation state is not supported by any experimental evidence and may require higher temperatures and O2 pressures than in our study38,39 but cannot be ruled out. Thus, it is suggested that the Cr2O3 surface active for HCl oxidation to Cl2 contains terminal Cr = O species of Cr5+ and Cr6+. With regards to the kinetics of the reaction steps, we expect the activation of gas-phase O2 by Cr3+ ions to produce oxidised surface chromium species to be rate limiting, as the HCl conversion was found to positively depend on the feed O2 concentration (Fig. 1b).
O) can form via dissociative adsorption of O2 on coordinatively-unsaturated Cr3+ cations, i.e. transforming Cr3+ into Cr5+ and completing its coordination shell.35–37 The generation of chromyl terminations with Cr in (VI) oxidation state is not supported by any experimental evidence and may require higher temperatures and O2 pressures than in our study38,39 but cannot be ruled out. Thus, it is suggested that the Cr2O3 surface active for HCl oxidation to Cl2 contains terminal Cr = O species of Cr5+ and Cr6+. With regards to the kinetics of the reaction steps, we expect the activation of gas-phase O2 by Cr3+ ions to produce oxidised surface chromium species to be rate limiting, as the HCl conversion was found to positively depend on the feed O2 concentration (Fig. 1b).
3.2. Supported Cr2O3
| Catalyst | State | X HCl (%) | S BET (m2 g–1) | Cr content (wt%) | Cr lossc (wt%) | Crystallite sizee (nm) | |
|---|---|---|---|---|---|---|---|
| 15 min | 3 h | ||||||
| a Conditions: O2/HCl = 2, Tbed = 678 K, P = 1 bar, W/ṅ0(HCl) = 5.6 g h mol−1. b Total surface area of the supports in brackets. c Determined after reaction for 3 h under the conditions in ‘a’. d Chromium content/loss after 40 h on stream in brackets. e Determined by application of the Scherrer equation to the XRD data. f Chromium content/loss at O2/HCl = 4, Tbed = 678 K, P = 1 bar, W/ṅ0(HCl) = 5.6 g h mol−1, and t = 10 h. | |||||||
| Cr2O3/SiO2 | Fresh | 28 | 26 | 149 (189)b | 13.4 | — | 27 |
| Used | — | — | 149 | 12.8 (9.7)d | 4.5 (27.6)d | 30 | |
| Used | — | — | — | 12.6f | 6.3f | — | |
| Cr2O3/Al2O3 | Fresh | 28 | 30 | 162 (191) | 12.7 | — | 8 |
| Used | — | — | 148 | 8.5 | 32.5 | 10 | |
| Used | — | — | — | 9.2f | 27.4f | — | |
| Cr2O3/TiO2 | Fresh | 21 | 14 | 34 (52) | 13.3 | — | 14 |
| Used | — | — | 32 | 12.8 | 3.7 | 16 | |
Fig. 6a shows the dependence of the rate of Cl2 production per gram of catalyst on the feed O2/HCl ratio over Cr2O3/SiO2. Data for bulk Cr2O3 have been included for comparative purposes. Cr2O3/SiO2 appears ca. 2 times more active than bulk Cr2O3 at O2/HCl = 7. If the rate is calculated per mol of chromium, the activity of Cr2O3/SiO2 exceeds that of bulk Cr2O3 by one order of magnitude at the same O2/HCl ratio. The rate increased upon raising the relative O2 content in the feed mixture and the formal reaction order on O2 for Cr2O3/SiO2 is 0.3 (±0.02). This dependence is thus stronger for the supported catalyst than for bulk Cr2O3 (reaction order = 0.2, Fig. 1b and 6a). Catalytic testing of Cr2O3/SiO2 at temperatures between 560 and 680 K at O2/HCl = 2 (Fig. 1d) enabled the estimation of Ea, which is 96 (±1.5) kJ mol−1. The value is practically identical to that obtained for unsupported Cr2O3 (97 kJ mol−1), suggesting that the kinetics of HCl oxidation is not affected by deposition of the active phase on this carrier.
 | ||
| Fig. 6 (a) Rate of Cl2 production per gram of catalyst versus feed O2/HCl ratio. (b) Stability test of Cr2O3/SiO2 and Cr2O3/Al2O3 showing the fraction of initial activity versus time-on-stream. (c) Deactivation constant and chromium loss in stability tests versus feed O2/HCl ratio. Conditions are detailed in Section 2.2. | ||
The long-term performance of Cr2O3/SiO2 was tested in a 40-h run at O2/HCl = 2. Fig. 6b shows the fraction of initial activity (F = Xt/X0) as a function of time for this catalyst, where Xt and X0 represent the HCl conversion at time t and 0 h, respectively. X0 was obtained by extrapolation of the linear trend line of the XHClversus time-on-stream plot. F consistently declined throughout the test, indicating strong and constant deactivation. Aiming at determining experimental conditions that would minimise this phenomenon, Cr2O3/SiO2 as well as Cr2O3/Al2O3 were tested at different feed O2/HCl ratios (between 0.5–7) for 10 h on stream. The deactivation constant, γ (h−1), was derived in each case from the slope of the trend lines (F = 1–γt) attained by plotting F versus t, as depicted in Fig. 6b. The resulting correlation between γ and the O2/HCl ratio is shown in Fig. 6c. For Cr2O3/SiO2, the value of γ linearly decreased raising the O2/HCl ratio up to 4, but a further increase in the O2/HCl ratio did not lead to better stabilisation. Consequently, O2/HCl = 4 was identified as the optimum feed ratio for this catalyst. For Cr2O3/Al2O3, γ was ca. 2 times higher than for Cr2O3/SiO2, indicating much stronger deactivation (Fig. 6c). Very high O2/HCl ratios, which would lie out of the ranges of economic operation conditions, would likely be required to curtail the activity loss. The origin of the observed deactivation is tackled in Section 3.3.
 | ||
| Fig. 7 XRD patterns of the supported Cr2O3 samples in fresh and used (in O2/HCl = 2 at 678 K for 3 h) forms. Reflections of Cr2O3 (JCPDS 70-3766) are indicated by (○). Unmarked reflections belong to the carriers. | ||
The H2-TPR profiles of fresh and used supported catalysts (Fig. 8) are characterised by two peaks due to chromium reduction, while the supports were irreducible under the applied conditions. The position of these features is substantially similar to the case of bulk Cr2O3. Thus, peak 1 is again attributed to the reduction of Cr6+ and/or Cr5+ to Cr3+ and peak 2 to the reduction of Cr3+ to Cr2+. Nevertheless, the variable degree of dispersion of the active phase, its particle size, and extent of interaction with the supports determined evident shifts of the peaks maxima.32 Still, in all of the cases their relative intensity reveals that a larger amount of chromium is present in higher oxidation states in the fresh catalysts, contrarily to the bulk oxide. This finding, combined with the greater exposed surface, likely explains the higher activity of supported with respect to bulk Cr2O3 (Fig. 6a). The intensities of peak 1 for the various catalysts are clearly different. For instance, Cr2O3/Al2O3 contains ca. 18 times more Cr5+/Cr6+ species than Cr2O3/SiO2. Hence, the properties of supports also determine in which chemical form chromium is stabilised on their surface. The anchoring process of chromium on metal oxides has been described as an acid–base type reaction, in which the weaker acid H2O is replaced by the stronger acid H2CrO4 (or H2Cr2O7).40 Typically, silica is more acidic than alumina. Consistently, the PZC of the SiO2 and Al2O3 employed in this study were measured as 3.4 and 8.0, respectively. Thus, mono-chromates are readily fixed onto the alumina surface in a well-dispersed manner, but a combination of mono- and poly-chromates would rather form on silica. In the latter case, the interaction with the surface hydroxyl groups of the support is insufficient to break the poly-chromate clusters into mono-chromates. In addition, silica usually offers fewer OH sites than alumina. This determines that a lower number of chromates can actually graft, while a larger fraction is only weakly attached.41 As a result, Cr6+ species are less dispersed and calcination leads to bigger Cr2O3 aggregates.40 XRD is most likely blind with respect to chromate species in view of their very small size. The prevalence of supported chromium species in either high or low oxidation state is corroborated by the appearance of the catalysts (Fig. 9). The dark orange colour of fresh Cr2O3/Al2O3, more similar to CrO3, suggests that it is rich in Cr6+ species, whereas the pale olive tone of Cr2O3/SiO2 resembles that of Cr2O3, pointing to the dominance of Cr3+ species in this catalyst. Considering the H2-TPR profiles of the used samples, peak 1 remarkably decreased in intensity, suggesting that Cr6+ species either volatilised or reduced to Cr3+ during reaction, as observed for bulk Cr2O3 (Fig. 3, profiles a, b). The peak was almost depleted for Cr2O3/Al2O3. The compositional change can be visualised by a clear alteration of the colour from dark orange to pale green (Fig. 9). Additionally, a coloured condensate was collected downstream the reactor in the first few minutes of the test (vide supra). Although this catalyst is expected to be more active than Cr2O3/SiO2 in view of the higher amount of Cr5+/Cr6+ species present, its instability likely caused substantial deactivation already before the first activity measurement. For Cr2O3/SiO2 the intensity of the H2-TPR peak 1 was about halved and the material assumed a somewhat darker green shade.
 | ||
| Fig. 8 H2-TPR profiles of the supported Cr2O3 samples in fresh and used (in O2/HCl = 2 at 678 K for 3 h) forms. | ||
 | ||
| Fig. 9 Pictures of the representative catalysts prior to and after reaction. | ||
3.3. Chromium loss and catalyst stability
Stability constitutes one essential parameter in catalyst development and is particularly critical in the case of HCl oxidation. Due to the health hazards associated with chromium, it is relevant to discuss the deactivation mechanism and, thus, if chromium is lost upon use and to which extent and in which form this occurs.Bulk Cr2O3 exhibited a stable activity in the short-time screening and no bulk chloride phases were detected by XRD in the used sample. Nevertheless, analysis of the zeolite material used as a guard bed during the test indicated a minor but measurable chromium loss (ca. 0.05 wt% during 5 h on stream). The performance of supported Cr2O3 catalysts has been later shown to strongly deteriorate already in a short time frame (10–40 h). Elemental analysis of the used supported catalysts confirmed chromium depletion in all cases (Table 2). Cr2O3/Al2O3 suffered from the highest chromium loss, ca. 32% after 3 h on stream. For SiO2- and TiO2-supported catalysts, the decrease in metal content was only ca. 4% upon the same treatment, but a similar value (28%) was attained for Cr2O3/SiO2 after 40 h on stream. Fig. 6c indicates that the variation of the deactivation constant and of the chromium loss at different O2/HCl ratios agrees well for Cr2O3/SiO2 and Cr2O3/Al2O3. This proves that the prime origin of activity deterioration of Cr2O3-based catalysts is the volatilisation of the active phase. The fact that no bulk chloride was detected after reaction suggests that the latter occurs via a different mechanism compared to copper-based catalysts,20 or that the instability of the chlorine-containing chromium phases possibly formed is much higher.
According to the literature, Cr2O3 (Cr3+) as well as surface chromyl species (Cr5+) produced during reaction should be stable at the reaction temperature,37 while Cr6+ species, namely, CrO3, CrO2(OH)2, and CrO2Cl2 are very labile.38,42–44 The contribution of CrO3 to the chromium loss is considered negligible in view of the reasons cited in Section 3.1.4 and its reactivity (herein explained). Therefore, the observed metal depletion is suggested to be mainly due to CrO2(OH)2 and/or CrO2Cl2. CrO2(OH)2 is generated by the action of O2 and H2O above 673 K from Cr3+38,44 and of H2O only from Cr6+ species between 408 and 458 K45 and, therefore, possibly formed also under our experimental conditions (eqn (1) and (2)). CrO2Cl2 can be produced as a gaseous species by the reaction of HCl with surface chromates already at 403 K (eqn (3)).42,43 This compound can either leave the solid or, upon interaction with surface hydroxyl groups, generate surface Cr(VI) monochlorides, which can decompose into chromates liberating Cl2 in the presence of O2.42 Hence, a higher feed O2 concentration should reduce the chromium loss related to the formation of CrO2Cl2. A significantly slower deactivation rate and lower metal loss were in fact observed when the reaction was performed in O2 excess (Fig. 6c), although the effect levelled off at a feed O2/HCl ratio above 4. Consequently, formation of CrO2Cl2 might be regarded as the dominant deactivation path. The higher chromium depletion for Cr2O3/Al2O3 can be rationalised in view of the presence of a larger initial amount of oxidised chromium species, compared to Cr2O3/SiO2 and Cr2O3/TiO2 (Section 3.2.2, Fig. 8), which can directly react with HCl or product H2O resulting in both CrO2Cl2 and CrO2(OH)2 (eqn (1) and (3)).42
| ½Cr2O3 + ¾O2 + H2O ↔ CrO2(OH)2 | (1) |
| CrO3 + H2O ↔ CrO2(OH)2 | (2) |
| CrO3 + 2HCl ↔ CrO2Cl2 + H2O | (3) |
The deactivation pathway of chromium-based catalysts offers a novel facet with regards to the activity loss of Deacon catalysts. In fact, it is neither based on bulk chlorination, as in the case of copper-based systems,1 nor on sintering, as for the ruthenium-based system,12 but rather proceeds through the formation of species (containing and/or non-containing chlorine) which are highly unstable under reaction conditions.
Taking into account the enormous activity drop, chromium loss, and, possibly, operational issues (mechanical instability of the catalyst shapes), chromium-based systems, in a fixed-bed configuration, prove not suitable for industrial application to HCl oxidation. Indeed, as mentioned in the Introduction, Mitsui suggested the use of a fluidised-bed reactor, the regular replenishment of the vaporised catalyst portion by a fresh load,7 and the addition of (allegedly) stabilizing compounds of potassium, copper, and lanthanum to the Cr2O3/SiO2 catalyst.46 Nevertheless, off gas and waste liquid streams have to be carefully treated to comply with the allowable limit of Cr(VI) (0.1 ppm).
4. Conclusions
Fundamental aspects of HCl oxidation to Cl2 on bulk and supported Cr2O3 catalysts, covering performance, stability, and mechanism, have been investigated. Bulk Cr2O3 resulted remarkably active and apparently stable. Kinetic experiments of this oxide as well as CrO3 in a fixed-bed reactor coupled to various physico-chemical characterisations provided explicit evidence for the active species and enabled us to derive a catalytic redox cycle for HCl oxidation, in which chromium reversibly cycles between the (III) and higher (V and VI) oxidation states. Oxidation of Cr2O3 by gas-phase O2 to generate surface chromyl species appeared as rate determining, owing to the positive dependence of the HCl conversion on the inlet oxygen content. The chemical, textural, and structural properties of the carriers (silica, alumina, and titania) greatly influenced the particle size and distribution as well as the dominant oxidation state of supported chromium. Cr2O3/SiO2, mainly featuring bigger particles and chromium in (III) oxidation state, exhibited superior performances. The kinetics of the partial O2 pressure and the dependence on the reaction temperature found for this catalyst were similar to those of bulk Cr2O3. Nevertheless, all supported catalysts underwent substantial chromium loss, ultimately resulting in dramatic activity deterioration. Among the various labile Cr6+ species possibly responsible for the metal depletion, CrO2Cl2 seems to play a predominant role, as suggested by the significant decrease in the deactivation rate at high inlet O2/HCl ratios. The short lifetime and the major chromium loss likely represent important reasons for the limited industrial success of chromium-based catalysts in HCl oxidation.Acknowledgements
We thank Bayer MaterialScience for granting permission to publish these results.References
- J. Pérez-Ramírez, C. Mondelli, T. Schmidt, O. F.-K. Schlüter, A. Wolf, L. Mleczko and T. Dreier, Energy Environ. Sci., 2011, 4, 4786 Search PubMed
.
-
H. Deacon, US85 370, assigned to Gaskell, Deacon and Co., 1868 Search PubMed
-
A. J. Johnson and A. J. Cherniavsky, US2 542 961, assigned to Shell Development Company, 1951 Search PubMed
- H. Y. Pan, R. G. Minet, S. W. Benson and T. T. Tsotsis, Ind. Eng. Chem. Res., 1994, 33, 2996 CrossRef CAS
- M. Mortensen, R. G. Minet, T. T. Tsotsis and S. Benson, Chem. Eng. Sci., 1999, 54, 2131 CrossRef CAS
-
T. Kiyoura, Y. Kogure, T. Nagayama and K. Kanaya, US4 822 589, assigned to Mitsui Toatsu Chemicals, 1989 Search PubMed
-
H. Itoh, Y. Kono, M. Ajioka, S. Takenaka and M. Kataita, US4 803 065, assigned to Mitsui Toatsu Chemicals, 1989 Search PubMed
- K. Seki, Catal. Surv. Asia, 2010, 14, 168 CrossRef CAS
- H. Ando, Y. Uchida, K. Seki, C. Knapp, N. Omoto and M. Kinoshita, Sumitomo Kagaku, 2010, 2, 1 Search PubMed
-
T. Hibi, H. Nishida and H. Abekawa, US5 871 707, assigned to Sumitomo Chemical Company, 1999 Search PubMed
-
A. Wolf, L. Mleczko, O. F. Schlüter and S. Schubert, EP2026905, assigned to Bayer MaterialScience, 2006 Search PubMed
- C. Mondelli, A. P. Amrute, F. Krumeich, T. Schmidt and J. Pérez-Ramírez, ChemCatChem, 2011, 3, 657 CrossRef CAS
- D. Crihan, M. Knapp, S. Zweidinger, E. Lundgren, C. J. Weststrate, J. N. Andersen, A. P. Seitsonen and H. Over, Angew. Chem., Int. Ed., 2008, 47, 2131 CrossRef CAS
- N. López, J. Gómez-Segura, R. P. Marín and J. Pérez-Ramírez, J. Catal., 2008, 255, 29 CrossRef
- M. A. G. Hevia, A. P. Amrute, T. Schmidt and J. Pérez-Ramírez, J. Catal., 2010, 276, 141 CrossRef CAS
- D. Teschner, R. Farra, L.-D. Yao, R. Schlögl, H. Soerijanto, R. Schomaecker, T. Schmidt, L. Szentmiklósi, A. P. Amrute, C. Mondelli, J. Pérez-Ramírez, G. Novell-Leruth and N. López, J. Catal., 2012, 285, 273 CrossRef CAS
- S. Zweidinger, D. Crihan, M. Knapp, J. P. Hofmann, A. P. Seitsonen, C. J. Weststrate, E. Lundgren, J. N. Andersen and H. Over, J. Phys. Chem. C, 2008, 112, 9966 CAS
- F. Studt, F. Abild-Pedersen, H. A. Hansen, I. C. Man, J. Rossmeisl and T. Bligaard, ChemCatChem, 2010, 2, 98 CrossRef CAS
- H. Over, J. Phys. Chem., 2012, 116, 6779 CrossRef CAS
- C. Mondelli, A. P. Amrute, T. Schmidt and J. Pérez-Ramírez, Chem. Commun., 2011, 47, 7173 RSC
- A. P. Amrute, C. Mondelli, M. Moser, G. Novell-Leruth, N. López, D. Rosenthal, R. Farra, M. E. Schuster, D. Teschner, T. Schmidt and J. Perez-Ramírez, J. Catal., 2012, 286, 287 CrossRef CAS
- A. G. Aglulin, Kinet. Catal., 1998, 39, 521 CAS
- A. P. Amrute, C. Mondelli, M. A. G. Hevia and J. Pérez-Ramírez, ACS Catal., 2011, 1, 583 CrossRef CAS
- G. C. Allen and P. M. Tucker, Inorg. Chim. Acta, 1976, 16, 41 CrossRef CAS
- G. C. Allen, S. J. Harris, J. A. Jutson and J. M. Dyke, Appl. Surf. Sci., 1989, 37, 111 CrossRef CAS
- B. Wichterlova, L. Krajcikova, Z. Tvaruskova and S. Beran, J. Chem. Soc., Faraday Trans. 1, 1984, 80, 2639 RSC
- A. P. Amrute, C. Mondelli, M. A. G. Hevia and J. Pérez-Ramírez, J. Phys. Chem. C, 2011, 115, 1056 CAS
- B. Grzybowska, J. Słoczyński, R. Grabowski, K. Wcisło, A. Kozłowska, J. Stoch and J. Zieliński, J. Catal., 1998, 178, 687 CrossRef CAS
- L. I. Ilieva and D. H. Andreeva, Thermochim. Acta, 1995, 265, 223 CrossRef CAS
- B. M. Weckhuysen, I. E. Wachs and R. A. Schoonheydt, Chem. Rev., 1996, 96, 3327 CrossRef CAS
- J. R. Sohn and S. G. Ryu, Langmuir, 1993, 9, 126 CrossRef CAS
- N. E. Fouad and H. Knözinger, Z. Phys. Chem., 1994, 186, 231 CrossRef CAS
- B. M. Weckhuysen and I. E. Wachs, J. Phys. Chem., 1996, 100, 14437 CrossRef CAS
- J. H. Uhm, M. Y. Shin, J. Zhidong and J. S. Chung, Appl. Catal., B, 1999, 22, 293 CrossRef CAS
- A. Zecchina, S. Coluccia, L. Cerruti and E. Borello, J. Phys. Chem., 1971, 75, 2783 CrossRef
- P. J. M. Carrott and N. Sheppard, J. Chem. Soc., Faraday Trans. 1, 1983, 79, 2425 RSC
- S. C. York, M. W. Abee and D. F. Cox, Surf. Sci., 1999, 437, 386 CrossRef CAS
- H. Asteman, J.-E. Svensson, L.-G. Johansson and M. Norell, Oxid. Met., 1999, 52, 95 CrossRef CAS
- H. C. Graham and H. H. Davies, J. Am. Ceram. Soc., 1971, 54, 89 CrossRef CAS
- B. M. Weckhuysen, R. A. Schoonheydt, J. Jehng, I. E. Wachs, S. J. Cho, R. Ryoo, S. Kijlstra and E. Poels, J. Chem. Soc., Faraday Trans., 1995, 91, 3245 RSC
- B. M. Weckhuysen, L. M. D. Ridder, P. J. Grobet and R. A. Schoonheydt, J. Phys. Chem., 1995, 99, 320 CrossRef CAS
- M. P. McDaniel, J. Catal., 1982, 76, 17 CrossRef CAS
- M. P. McDaniel, J. Catal., 1982, 76, 29 CrossRef CAS
- E. J. Opila, D. L. Myers, N. S. Jacobson, I. M. B. Nielsen, D. F. Johnson, J. K. Olminsky and M. D. Allendorf, J. Phys. Chem. A, 2007, 111, 1971 CrossRef CAS
- O. Glemser and A. Müller, Z. Anorg. Allg. Chem., 1964, 334, 150 CrossRef CAS
-
K. Miyata, J. Morisaki, T. Hirayama, H. Kamachi and K. Yamada, EP0711599A1, assigned to Mitsui Toatsu Chemicals, 1996 Search PubMed
| This journal is © The Royal Society of Chemistry 2012 |