Electro-oxidation of formic acid using polyindole-SnO2 nanocomposite
Received
6th June 2012
, Accepted 11th August 2012
First published on 14th August 2012
Abstract
We report a chemical oxidative method for polyindole-tinoxide (Pin–SnO2) synthesis and its electro-catalytic application. Our method consists of chemical polymerization of an indole monomer in the presence of dispersed nano SnO2 at two different pHs i.e. below and above the isoelectric point of the SnO2. Nano SnO2 with crystallite sizes of nearly 5.0 nm are prepared by precipitation technique. For the sake of comparison, pristine polyindole is also synthesized by the same chemical method. These materials are characterized by XRD, FTIR and TEM. Characterization confirms the presence of nano SnO2 particles in a Pin matrix and the effect of pH on the formation of a uniform nanocomposite and the extent of interaction of fillers and matrix. The electro-catalytic properties of the Pin–SnO2 nanocomposite are investigated for formic acid oxidation using the voltammetric technique. This shows excellent synergic effect for electro-catalytic activity of the low doped Pin–SnO2 nanocomposite (Pin–SnO2 nanocomposite formed at higher pH). This composite is well suited for CO tolerance during the oxidation of formic acid in acidic medium and follows a direct pathway to convert it into CO2.
1. Introduction
Formic acid (FA, HCOOH) is a potentially attractive fuel for fuel cell technology not only because of better proton transport on the catalyst layer with a high thermodynamic cell voltage, but also due to it being more environmentally friendly and non-explosive in its diluted form than methanol.1–3 It has been reported that the electro-oxidation of FA undergoes a triple path mechanism that starts with the adsorption of formic acid. After that, the adsorbent can either be directly oxidized to carbon dioxide (direct pathway) or dehydrogenated to bridge-adsorbed as formates (formate pathway) or undergo dehydration to adsorbed carbon monoxide (indirect pathway) as shown below:4
| Direct pathway: HCOOH + S → CO2 + 2H+ + 2e− |
| Formate pathway: HCOOH + S → S–OOCH + H+ +e− |
| Indirect pathway: HCOOH + S → S–CO + H2O |
| S–CO + Me–OH → S + CO2 + H+ + e− |
where ‘S’ is the substrate, like Pt, Pd, TiO2 or composites.
The direct pathway is the perfect pathway, because for the direct pathway, formic acid is directly oxidized to CO2 and no poisonous CO intermediate is formed. Moreover, self poisoning of pure metal electrodes limits their properties. Various reports are available for formic acid oxidation using metals (e.g. Pt, Pd, Au), bimetals (e.g. Fe–Pt, Pt–Pd), metal polymers, metal oxides (TiO2, PbO, CeO2), and metal oxide based polymers.5–16 Among these catalysts, Pt metal shows a dual pathway mechanism (i.e. direct and indirect oxidation pathway) of formic acid.17–19 To overcome this behavior, Pt based composites are studied for the direct oxidation of formic acid, but their preparation processes are costly due to complicated steps of synthesis.20–22 It is desirable to develop easier and cheaper routes. On the other hand, Pd and Au are studied as well suited catalysts for the direct oxidation of formic acid.23,24 However, the corrosion of the metallic catalyst in the presence of acidic or basic media and CO-poisoning by FA limit their usage as electrode materials.18,19,25,26 Metal oxide or metal oxide based composites are better ways to overcome these limitations.
It has been observed that the catalysts (mainly as the fillers) in the case of composite materials at the anode electrode impact on the overall performance of cell. For the stable and efficient performance of a fuel cell these materials are crucial as they may affect the charge conductivity, catalyst dispersion and stability.14,21,27
In metal oxides, tin oxide (SnO2) is found to be an interesting material as a catalyst (as the filler in composites) due to its dual valency (+II and +IV), which facilitates variation in the surface oxygen composition.28 Due to this property it has unique physicochemical properties, especially for oxygen containing compounds, as well as being quite stabile in dilute acid solution.29 It has been suggested that SnO2 itself could be widely used for CO-oxidation.30 However, for a good electro-catalyst, CO tolerance capability, low cost, cheaper synthetic process and remarkable electronic conductivity of electro-catalytic materials are important parameters. Therefore, for the SnO2 catalyst, CO tolerance capability and electronic conductivity need to improve. One of the simple methods may be the use of a conductive matrix having good redox properties, like conducting polymers. Heterocyclic conducting polymers are excellent candidates in this context because of their good stability and participation of the hetero-atom during redox performance.31 However, their swelling property in aqueous media limits their application.32 Among the heterocyclic conducting polymers, polyindole (Pin) has attracted considerable interest due to the combinational properties of both poly(p-phenylene) and polypyrrole together,33 such as fairly good thermal stability, high stability and redox property, slow degradation and better air stability.34–36 These properties make the Pin suitable for potential applications in diversified fields as sensor and biosensor, catalyst, fuel cell batteries etc.21,37–39
In view of the above we tried to explore the nanocomposite of SnO2 and Pin for electro-catalysis of FA. To the best of our knowledge, up to now there has been no report on performance of Pin–SnO2 nanocomposite based catalysts for the direct oxidation of FA. In the present work, the Pin–SnO2 nanocomposite has been successfully synthesized by a chemical method at two different pHs (above and below the isoelectric point of the SnO2). The as-prepared nanocomposites were characterized by XRD, FTIR and TEM. Electrochemical technique was employed for the demonstration of FA oxidation over nanocomposite modified glassy carbon electrode (GCE) in a mixture of 1.0 M HCOOH + 0.5 M H2SO4 as electrolyte.
2. Experimental
2.1 Materials
Stannous chloride and Ammonium persulphate were purchased from HiMedia, India, Ethanol, Ammonia solution and Sulphuric acid were purchased from Merck, India. Indole was purchased from Aldrich, USA.
2.2 Synthesis of nano SnO2
Tin oxide nanoparticles were synthesized by the precipitation technique using following procedure:
1.43 g SnCl4·5H2O was added to 20 mL of ethanol in a round bottom flask and stirred for 2 h. After that 25% ammonia solution was added dropwise with the help of burette under constant stirring at RT till the pH 9. Then, the white precipitate was filtered till the pH of the filtrate became neutral and then calcined at 400 °C for 2 h.
2.3 Synthesis of Pin–SnO2 nanocomposite
Two types of Pin–SnO2 nanocomposites were synthesized by varying the pH of the aqueous solvents. In this synthetic process 5 mg of nano SnO2 was dispersed in 20 mL of H2SO4 solution (pH 3). After that freshly prepared indole monomer solution (0.01 M) in ethanol was added dropwise to the SnO2 dispersion solution and polymerization was carried out by APS oxidizing agent (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 molar ratio w.r.t. monomer) in 24 h. The resulting Pin–SnO2 nanocomposite was collected by centrifugation followed by washing with Milli-Q water (finally with ethanol) and dried in vacuum oven at 50 °C. A similar procedure was adopted for the synthesis of the other Pin–SnO2 nanocomposite in H2SO4 solution (pH 6).
:2 molar ratio w.r.t. monomer) in 24 h. The resulting Pin–SnO2 nanocomposite was collected by centrifugation followed by washing with Milli-Q water (finally with ethanol) and dried in vacuum oven at 50 °C. A similar procedure was adopted for the synthesis of the other Pin–SnO2 nanocomposite in H2SO4 solution (pH 6).
2.4 Electrode fabrication
The construction of the Pin–SnO2 nanocomposite film on the surface of the GCE was performed as follows: 0.5 mg of Pin–SnO2 powdered nanocomposite material was mixed in 500 μL of THF and dispersed using ultrasonication for 15 min. Next a 5 μL aliquot of this suspension was pipetted onto the surface of freshly polished GCE. For comparison purposes, a nano SnO2 and a Pin modified GCE were also prepared in the same manner.
2.5 Instrumentation
The as-synthesized nano SnO2, Pin and Pin–SnO2 nanocomposites were analyzed for structural properties and electrochemical oxidation of FA using various characterization techniques. UV-vis spectroscopy was performed on a Perkin Elmer UV-vis spectrophotometer (Model Lambda-25; Germany). XRD was performed on a Rigaku D/max-2200PC diffractometer using Cu Kα radiation with a wavelength of 1.54 Å. FTIR spectra were recorded on a Nicolet-6700, USA (32 scans at a resolution of 4 cm−1). The samples were pelleted with dried KBr and analyzed in transmission mode. Transmission electron microscopy (TEM) was performed with an FEI, Tecnai 30 G2S-Twin electron microscope, USA, at an operating voltage of 300 kV (courtesy of Nanotechnology Application Centre, University of Allahabad). The samples were diluted in ethanol and a drop was deposited onto a carbon-coated grid.
Differential pulse voltammetry (DPV) and Chronoamperometry (CA) were performed on a CH-Instrument Electrochemical Workstation (Model 7401; USA) and Metrohm Autolab (PG STAT-12/30/302; The Netherlands) using a three electrode system. In this technique Ag/AgCl as the reference, Pt-plate as the counter and glassy carbon modified by Pin–SnO2 nanocomposites (Pin–SnO2/GC) as the working electrode were used to investigate the FA oxidation in a mixture of 1.0 M HCOOH + 0.5 M H2SO4 as the electrolyte.
3. Results and discussion
The isoelectric point is the important parameter to explain the overall acid–base behavior of metal oxide surfaces. In this system there is a competition between the acidity of the metal cation and the basicity of oxygen.40 For example, the isoelectric point of SnO2 is 4–5, in which surface speciation is dominated by the protonated bridging group (Sn2OH+) and deprotonated terminal hydroxide group (SnOH−) by varying pHs.41,42 At low pH, the SnO2 clusters behave as positively charged moieties due to the adsorption of H+ ions over the surface oxygen.43 While at high pH, the SnO2 clusters behave as negatively charged moieties due to adsorption of OH− ions (aqueous) or HOH over the metal.42 Apart from this, pH alteration of the SnO2 suspension affects the overall particle sizes. The particle sizes get enlarged on decreasing pH due to collision and coalescence phenomenon as reported by other workers also.44 It has been reported that macrocyclic compounds and electron rich compounds (like indole, Pin, polycarbazole) create a synergic effect by donating their available electrons to the electron deficient clustered compound.14,21,45,46 However, in our case, at low pH the Pin is highly oxidized in the vicinity of the SnO2 clusters due to the exchange of protons to the polymer (low pH caused doping on Pin). While polymerization occurs at high pH, Pin is not highly oxidized in the vicinity of the SnO2 cluster. Thus, probably the electron is more localized (in the latter case) over the hetero atom of the Pin and it is efficient to donate its electron to the electron deficient SnO2 clusters. This charge transfer is assumed on the basis of interaction between the organic molecules and metal oxide clusters, however, no evidence of any primary bonding was observed. In view of this, the nanocomposite formed at high pH may be better over the low pH formed nanocomposite. Both of the nanocomposites were characterized and studied for the electro-catalytic application.
UV-vis analysis
UV-vis absorption spectra of the nano SnO2, pure Pin and Pin–SnO2 nanocomposites (formed at pH 3 and 6), recorded at room temperature in DMSO solvent, are shown in Fig. 1. For nano SnO2, the narrow absorption onset with a peak at 275 nm suggested the direct transition of electrons and narrow size distribution in particles that can also be seen in the TEM image, which is explained latter.47 The as-synthesized pure Pin and Pin–SnO2 nanocomposites change their colour from green (oxidized state polymer) to light brown (at pH 6) and dark brown (at pH 3) (cf. inset of Fig. 1).
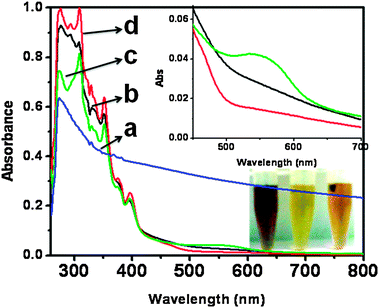 |
| | Fig. 1 UV-vis spectra of (a) nano SnO2 (b) pure Pin, (c) and (d) Pin–SnO2 nanocomposites (formed at pH 3 and pH 6, respectively). Inset: Enlarged view of 275 nm to 375 nm for Pin (black), the Pin–SnO2 nanocomposite at pH 3 (green) and the Pin–SnO2 nanocomposite at pH 6 (red). | |
Pure Pin and the Pin–SnO2 nanocomposites showed well known characteristic peaks (from 275–375 nm) due to π–π* and n–π* transitions.48,49 However, in the case of the Pin–SnO2 nanocomposite formed at pH 3, the decrease in peak intensity at 275 nm and an extra peak between 500–600 nm is due to an increase in doping of the corresponding Pin.50,51
XRD analysis
The powered XRD pattern of the as-synthesized nano SnO2, pure Pin and Pin–SnO2 nanocomposites at different pHs are shown in Fig. 2. For nano SnO2, the presence of diffraction peaks at plane 110, 101, 200, 211, 220, 002, 112 and 301 (cf.Fig. 2a) suggested a pure rutile type structure having cassiterite phase.47 This pattern is consistent to JCPDS (card no. 41-1445). However, these diffraction peaks are broadened due to small particle size. In fact, the crystallite sizes are calculated as 5.11 nm using the Debye–Scherrer equation.52 In this regard, the spectral data was deconvoluted using Lorentzian fitting, which is shown in the inset of Fig. 2. The XRD pattern of pure Pin (cf.Fig. 2d) consists of a distinctive broad peak at 2θ = 19° and 27° suggesting amorphous nature.53 The presence of planes 110, 101, 200, 211 and 112 in the Pin–SnO2 nanocomposites (formed at pH 3 and 6; cf.Fig. 2b and c) are evidence of nano SnO2 intercalation in the Pin matrix. The presence of nano SnO2 particles inside the polymer matrix can also be observed by TEM.
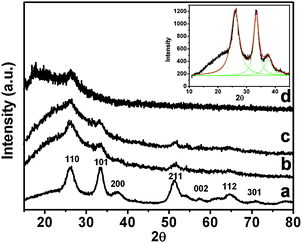 |
| | Fig. 2 XRD of (a) nanostructured SnO2, (b) and (c) Pin–SnO2 nanocomposites at pH 3 and 6, respectively, and (d) pure Pin. Inset: the Lorentzian fitting for nanostructured SnO2. | |
FTIR analysis
The structural features of nano SnO2, pure Pin and Pin–SnO2 nanocomposites were characterized by FTIR analysis as shown in Fig. 3. The FTIR spectra of the as-synthesized nano SnO2 particles (as shown in Fig. 3a) are similar to that of nano SnO2 particles synthesized by others.54,55 The bands at 500 and 660 cm−1 are due to vibration of the O–Sn–O bridging bond and Sn–O terminal bond, respectively. The bands at 4310 and 1633 cm−1 are due to O–H stretching and the deformation vibration of weakly-bound water. All characteristic bands of Pin (shown in Fig. 3d) are similar to our earlier reported paper.56 However, in the Pin–SnO2 nanocomposites, the presence of characteristic bands of both the components (i.e. for nano SnO2 and Pin as shown in Fig. 3b and c) are clear evidence for the existence of nano SnO2 within the Pin matrix. However, the peaks for the O–Sn–O bridging bond and the Sn–O terminal bond were diminished significantly for the Pin–SnO2 formed at high pH.
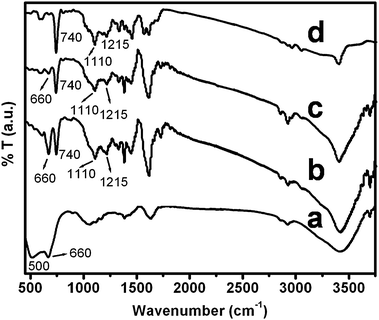 |
| | Fig. 3 FTIR spectra of (a) nano SnO2, (b) and (c) SnO2–Pin nanocomposites synthesized at pH 3 and pH 6, respectively, and (d) Pin. | |
TEM analysis
The TEM image of the agglomerated spherical particles of the as-synthesized nano SnO2 (with particle size = 5.5 nm) and the selected area diffraction pattern (SADP) are shown in Fig. 4a and b, respectively. The SADP of these nanoparticles is again characteristic of the rutile structure of their cassiterite phase. From the TEM image (Fig. 4a) it can be observed that there are two types of crystallites grown. The spherical and elongated crystallites growth supported that the particle growth occurred by a grain rotation induced grains coalescence mechanism, as has also been observed earlier.57,58 In the Pin–SnO2 nanocomposites, nano SnO2 particles are embedded individually within Pin matrix, which are visible in the TEM images (as shown in Fig. 4c and d), are another evidence for the existence of these particles inside the Pin matrix even with the variation of pHs. In these micrographs it can be also observed that nano SnO2 particles are surrounded by the Pin polymer.
 |
| | Fig. 4 (a) TEM of nano SnO2, (b) SADP of nano SnO2, (c) and (d) TEM of Pin–SnO2 nanocomposites synthesized at pH 3 and at pH 6, respectively. | |
Catalytic activity
The catalytic activity of the Pin–SnO2 nanocomposites was studied for electro-oxidation of FA and compared with bare GCE, pure Pin and nano SnO2.Fig. 5 shows the differential pulse voltammetry (DPV) for the bare GCE, pure Pin, nano SnO2 and the Pin–SnO2 nanocomposites (formed at two different pHs) in 1.0 M HCOOH + 0.5 M H2SO4. Two well known distinct oxidation peaks (0.36 V and 0.58 V vs. Ag/AgCl) are observed for FA at all the electrodes (however, peak current differs). The well defined sharp peak at 0.36 V is due to the oxidation direct pathway while the other small shoulder peak at 0.58 V is due to oxidation via indirect pathway59 (as also discussed in the introduction section). In the cases of Pin, the bare GCE, and nano SnO2 (cf.Fig. 5(i), (ii) and (iii)), both the oxidation peaks appeared in the range of 0.34 V to 0.66 V vs. Ag/AgCl, which indicates that the oxidation is following both direct and indirect pathways. However, for the Pin–SnO2 nanocomposites (as shown in Fig. 5(iv) and (v)), the profile indicates enhanced peak current at 0.34 V and stronger poisoning-tolerance ability for FA oxidation (direct pathway) in comparison to Pin or even nano SnO2. Further, it can be seen that the nanocomposite consisting of low doped Pin (which is formed at pH 6) suppressed the indirect pathway of FA oxidation more than the other nanocomposite consisting of high doped Pin (which is formed at pH 3). This may be due to the synergic effect of the Pin (low doped) matrix, similar to polycarbazole reported earlier,14 which reduces the CO formation over the substrate. In addition, the oxidation peak potential for the direct pathway of FA is much lower in the case of the Pin–SnO2 nanocomposite electrodes in comparison to nano SnO2.
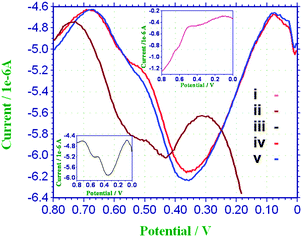 |
| | Fig. 5 DPV of (i) Pin (inset top), (ii) nano SnO2 particles (iii) GCE (inset bottom), (iv) and (v) Pin–SnO2 nanocomposites (formed at pH 3 and pH 6, respectively) in 1.0 M HCOOH + 0.5 M H2SO4. | |
The stability and electroactivity of the prepared samples nano SnO2, Pin and the Pin–SnO2 nanocomposites (formed at pH 3 and pH 6) were examined by chronoamperogram (CA) analysis. The CA is recorded by maintaining the electrode at a constant potential of 0.4 V or 0.6 V vs. Ag/AgCl for 300 s in a solution containing 0.1 M HCOOH + 0.5 M H2SO4, as shown in Fig. 6. In the general interpretation, a gradual decrease in current density per unit time is an indication of good electrode activity and high stability.60 At an applied potential of 0.4 V (vs. Ag/AgCl), the Pin–SnO2 nanocomposite formed at pH 6 exhibits the highest current density compared to the nano SnO2, Pin and Pin–SnO2 nanocomposite formed at pH 3. In the experiment, the current density for FA oxidation of the Pin–SnO2 nanocomposite formed at pH 6 gradually decreases up to 2.95 μA cm−2 at 300 s. In contrast, the current density in nano SnO2, Pin and the Pin–SnO2 nanocomposite formed at pH 3 decreases suddenly within 5 s and is observed as 0.5, 1.5 and 2.34 μA cm−2, respectively, at 300 s. The sudden decrease in the oxidation current densities on SnO2, Pin and the Pin–SnO2 nanocomposite formed at pH 3 might be due to CO poisoning, whereas the gradual decrease in current density of the Pin–SnO2 nanocomposite formed at pH 6 indicates the better resistivity toward CO poisoning.61 Moreover, at 0.6 V (vs. Ag/AgCl), we observed a similar trend as at 0.4 V, but relatively high current density is observed in the Pin–SnO2 nanocomposite formed at pH 6 compared to the other samples (Table 1). However, on comparing both results obtained at 0.4 V and 0.6 V, the Pin–SnO2 nanocomposite formed at pH 6 exhibits better CO tolerance at the higher potential (because it has high current density among the others), while the results for SnO2, Pin and the Pin–SnO2 nanocomposite formed at pH 3 support CO poisoning at 0.6 V.
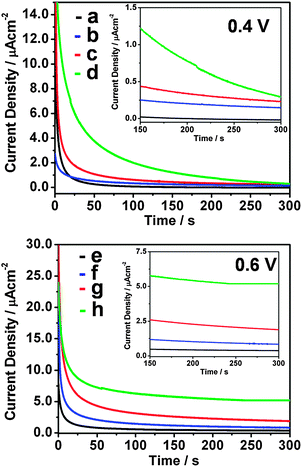 |
| | Fig. 6 CA of nano SnO2, pure Pin and the Pin–SnO2 nanocomposites formed at pH 3 and pH 6 (a)–(d) at 0.4 V vs. Ag/AgCl and (e)–(h) at 0.6 V vs. Ag/AgCl. Inset: enlarged view of the same CA from 150–300 s. | |
Table 1 Current density for FA oxidation for catalysts during CA analysis
| Materials |
Current density (in μA cm−2) |
| 0.4 V vs. Ag/AgCl |
0.6 V vs. Ag/AgCl |
| SnO2 |
0.50 |
0.40 |
| Pin |
1.50 |
0.80 |
| Pin–SnO2 (pH 3) |
2.34 |
1.88 |
| Pin–SnO2 (pH 6) |
2.95 |
5.20 |
4. Conclusion
In this paper, Pin–SnO2 nanocomposites have been prepared by chemical oxidation method and used for the electro-oxidation of formic acid in 0.5 M H2SO4 solution. Compared with their counterparts (i.e., Pin and nano SnO2 deposited on the GC and GCE alone), the Pin–SnO2 nanocomposites exhibit much better catalytic activity for FA electro-oxidation. The enhanced electro-catalytic performance mainly comes from the synergetic effect between Pin and nano SnO2. At the same time, it is found that the Pin–SnO2 nanocomposite consisting of low doped Pin (prepared at higher pH) has a better CO tolerance ability than the Pin–SnO2 nanocomposite consisting of high doped Pin (prepared at low pH). This result indicates that the low doped Pin–SnO2 nanocomposite can be used as an efficient promoter for the electro-oxidation of FA in comparison to conventional electrodes based on carbon or SnO2 or nano SnO2 catalysts.
References
- T. T. Cheng and E. L. Gyenge, J. Appl. Electrochem., 2009, 39, 1925 CrossRef CAS.
- C. Liao, Z. D. Wei, S. G. Chen, L. Li, M. B. Ji, Y. Tan and M. J. Liao, J. Phys. Chem. C, 2009, 113, 5705 CAS.
- W. Xu, Y. Gao, T. Lu, Y. Tang and B. Wu, Catal. Lett., 2009, 130, 312 CrossRef CAS.
- Y. X. Chen, M. Heinen, Z. Jusys and R. J. Behm, Angew. Chem., Int. Ed., 2006, 45, 981 CrossRef CAS.
- R. Zhou, W. Zhou, H. Zhang, Y. Du, P. Yang, C. Wang and J. Xu, Nanoscale Res. Lett., 2011, 6, 381 CrossRef.
- H. Miyake, T. Okada, G. Samjeske and M. Osawa, Phys. Chem. Chem. Phys., 2008, 10, 3662 RSC.
- R. K. Pandey and V. Lakshminarayanan, J. Phys. Chem. C, 2009, 113, 21596 CAS.
- V. Mazumder, M. Chi, M. N. Mankin, Y. Liu, O. Metin, D. Sun, K. L. More and S. Sun, Nano Lett., 2012, 12, 1102 CrossRef CAS.
- C. Liao, Z. D. Wei, S. G. Chen, L. Li, M. B. Ji, Y. Tan and M. J. Liao, J. Phys. Chem. C, 2009, 113, 5705 CAS.
- H. J. Yun, H. Lee, J. B. Joo, W. Kim and J. Yi, J. Phys. Chem. C, 2009, 113, 3050 CAS.
- A. Cuesta, G. Cabello, C. Gutierreza and M. Osawab, Phys. Chem. Chem. Phys., 2011, 13, 20091 RSC.
- M. Jin, H. Zhang, Z. Xie and Y. Xia, Energy Environ. Sci., 2012, 5, 6352 CAS.
- S. Murugesan, K. Myers and V. Subramanian, Appl. Catal., B, 2011, 103, 266 CrossRef CAS.
- W. Zhou, J. Xu, Y. Du and P. Yan, Int. J. Hydrogen Energy, 2011, 36, 1903 CrossRef CAS.
- W. Chen, J. Kim, S. Sun and S. Chen, Phys. Chem. Chem. Phys., 2006, 8, 2779 RSC.
- O. Winjobi, Z. Zhang, C. Liang and W. Li, Electrochim. Acta, 2010, 55, 4217 CrossRef CAS.
- Z. Z. Zhu, Z. Wang and H. L. Li, Appl. Surf. Sci., 2008, 254, 2934 CrossRef CAS.
- V. Selvaraj, M. Alagar and K. S. Kumar, Appl. Catal., B, 2007, 75, 129 CrossRef CAS.
- V. Selvaraj, A. N. Grace and M. Alagar, J. Colloid Interface Sci., 2009, 333, 254 CrossRef CAS.
- F. Ren, W. Zhou, Y. Du, P. Yang, C. Wang and J. Xu Inter, Int. J. Hydrogen Energy, 2011, 36, 641 CrossRef.
- W. Zhou, Y. Dua, H. Zhang, J. Xu and P. Yang, Electrochim. Acta, 2010, 55, 2911 CrossRef CAS.
- J. Xiang, R. Chenitz and J. P. Dedelet, J. Electrochem. Soc., 2010, 157, B1658 CrossRef.
- B. L. Wu and S. Chen, J. Electroanal. Chem., 2001, 517, 95 CrossRef.
- X. Wang, Y. Tang, Y. Gao and T. Lu, J. Power Sources, 2008, 175, 784 CrossRef CAS.
- V. A. Lavrenko, A. G. Koval, S. P. Gordienko, E. S. Lugovskaja, B. V. Fenochka and Yu. A. Klimovskii, Corros. Sci., 1978, 18, 809 CrossRef CAS.
- X. Gao, J. Tang, Y. Zuo, Y. Tang and J. Xiong, Corros. Sci., 2009, 51, 1822 CrossRef CAS.
- B. Zhang, D. Ye, J. Li, X. Zhu and Q. Liao, J. Power Source, 2012, 214, 277 CrossRef CAS.
- M. Batzill and U. Diebold, Prog. Surf. Sci., 2005, 79, 47 CrossRef CAS.
- A. G. Victoria and F. C. David, Surf. Sci., 1994, 312, 106 CrossRef.
- K. Grass and H. G. Lintz, J. Catal., 1997, 172, 446 CrossRef CAS.
- G. R. Hutchison, M. A. Ratner and T. J. Marks, J. Phys. Chem. B, 2005, 109, 3126 CrossRef CAS.
- G. Karthikeyan, S. Sahoo, G. C. Nayak and C. K. Das, Macromol. Res., 2011, 20, 351 CrossRef.
- C. Zhijiang and Y. Guang, Synth. Met., 2010, 160, 1902 CrossRef.
- D. Billaud, E. B. Maarouf and E. Hannecart, Synth. Met., 1995, 69, 571 CrossRef CAS.
- E. B. Maarouf, D. Billaud and E. Hannecart, Mater. Res. Bull., 1994, 29, 637 CrossRef CAS.
- P. S. Abthagir, K. Dhanalakshmi and R. Saraswathi, Synth. Met., 1998, 93, 1 CrossRef CAS.
- P. C. Pandey, D. S. Chauhan and R. Prakash, J. Appl. Polym. Sci., 2012, 125, 2993 CrossRef CAS.
- A. Kumar and R. Prakash, Chem. Phys. Lett., 2011, 511, 77 CrossRef CAS.
- G. Rajasudha, L. M. Jayan, D. Durgalakshmi, P. Thangadurai, N. Boukos, V. Narayanan and A. Stephen, Polym. Bull., 2011, 68, 181 CrossRef.
- M. Delamar, J. Electron Spectrosc. Relat. Phenom., 1990, 53, C11 CrossRef CAS.
- A. Kay and M. Gratzel, Chem. Mater., 2002, 14, 2930 CrossRef CAS.
- J. Rosenqvist, M. L. Machesky, L. Vlcek, P. T. Cummings and D. J. Wesolowski, Langmuir, 2009, 25, 10852 CrossRef CAS.
- A. C. Ibarguen, A. Mosquera, R. Parra, M. S. Castro and J. E. Rodriguez-Paez, Mater. Chem. Phys., 2007, 101, 433 CrossRef.
- C. Ribeiro, E. J. H. Lee, T. R. Giraldi, E. Longo, J. A. Varela and E. R. Leite, J. Phys. Chem. B, 2004, 108, 15612 CrossRef CAS.
- X. Zhou, C. Liu, J. Liao, T. Lu and W. Xing, J. Power Sources, 2008, 179, 481 CrossRef CAS.
- W. Zhoua, C. Wang, J. Xua, Y. Duc and P. Yang, J. Power Sources, 2011, 196, 1118 CrossRef.
- W. Chen, D. Ghosh and S. Chen, J. Mater. Sci., 2008, 43, 5291 CrossRef CAS.
- O. Eraldemir, B. Sari, A. Gok and H. I. Unal, J. Macromol. Sci., Part A: Pure Appl. Chem., 2008, 45, 205 CrossRef CAS.
- S. An, T. Abdiryim, Y. Ding and I. Nurulla, Mater. Lett., 2008, 62, 935 CrossRef CAS.
- K. Dhanalakshmi and R. Saraswathi, J. Mater. Sci., 2001, 36, 4107 CrossRef CAS.
- G. Nie, L. Zhou and H. Yang, J. Mater. Chem., 2011, 21, 13873 RSC.
- F. W. Jones, Proc. R. Soc. London, Ser. A, 1938, 166, 16 CrossRef CAS.
- N. B. Taylan, B. Sari and A. H. I. Unal, J. Polym. Sci., Part B: Polym. Phys., 2010, 48, 1290 CrossRef CAS.
- G. Zhang and M. Liu, J. Mater. Sci., 1999, 34, 3213 CrossRef CAS.
- Y. Wang, X. Wu, Y. Li and Z. Zhou, Solid-State Electron., 2004, 48, 627 CrossRef CAS.
- L. Joshi and R. Prakash, Mater. Lett., 2011, 65, 3016 CrossRef CAS.
- E. R. Laite, T. R. Giraldi, F. M. pontes, E. Longo, A. Beltran and J. Andres, Appl. Phys. Lett., 2003, 83, 1566 CrossRef.
- D. Moldovan, V. Yamakov, D. Wolf and S. R. Phillport, Phys. Rev. Lett., 2002, 89, 206101 CrossRef.
- O. Winjobi, Z. Zhang, C. Liang and W. Li, Electrochim. Acta, 2010, 55, 4217 CrossRef CAS.
- R. S. Jayashree, J. S. Spendelow, J. Yeom, C. Rastogi, M. A. Shannon and P. J. A. Kenis, Electrochim. Acta, 2005, 50, 4674 CrossRef CAS.
- J. Shim, J. Lee, Y. Ye, J. Hwang, S. K. Kim, T. H. Lim, U. Wiesner and J. Lee, ACS Nano DOI:10.1021/nn301692y.
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here.